By applying this logic, the three complementation groups are:
- r1 and r3
- r2, r5, and r6
- r4, r7, and r8.
Again, in order to get complementation (i.e., to get wild type phenotype when crossing mutations in two separate genes) the mutant phenotype has to be recessive -- if the mutant phenotype is dominant, we wouldn't see the wild type phenotype even in the heterozygotes. Therefore, none of these mutations had a dominant phenotype.
We can come up with a working hypothesis (i.e., a hypothesis that we'll work with, and modify or discard if need be) by looking at the various phenotypes.
First, the normal phenotype has brown and white coat color; there is no black coat. Therefore, the presence of black in a mutant suggests (but does not prove) the following: black color is normally converted to something else (such as brown or white), but that conversion has failed in the mutant. Since the mutation that gives black coats is dd, the wild type gene D is presumably (in this hypothesis) responsible for converting black to something else:
- D
- black ---------> (something else)
Furthermore, since D is epistatic to the other genes (i.e., in the absence of gene D, we get a black coat phenotype, regardless of the genotype of the other two genes), we can hypothesize that the action of gene D must be requied early in the pathway, before the action of genes B and E.
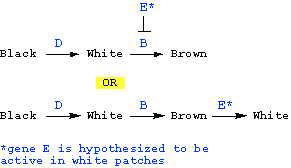
What is that "something else" that brown is converted to? The simplest hypothesis is that that something else is either white or brown. Let us consider each of these two possibilities:
Possibility 1: Black is converted to white.
In this case, we'd have to hypothesize further that gene B is required to convert white to brown pigment -- since the phenotype of bb mutants is fully white (i.e., no brown). What is role of gene E in this hypothesis? Since the wild type phenotype is brown and white patches, we'd have to say that the role of gene E in this hypothesis is either to block the action of gene B in some patches (so that those patches remain white, and are not converted to brown), or to reverse the effect of gene B in some patches (brown is converted back to white in those patches):
However this hypothesis predicts that B should be epistatic to E -- i.e., in the absence of wild type gene B, the phenotype should be fully white, regardless of whether gene E is wild type or mutant. However, looking at the given phenotypes, we see that this prediction is NOT met -- B is NOT epistatic to E (i.e., the phenotype of bb is not the same as bbee). Therefore, this hypothesis is NOT CONSISTENT with the data.
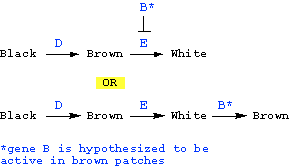
Possibility 2: Black is converted to brown.
Here, we'd hypothesize that gene E is required to convert brown pigment to white -- since the phenotype of ee mutants is fully brown (i.e., no white). What is role of gene B in this hypothesis? Since the wild type phenotype is brown and white patches, we'd have to say that the role of gene B in this hypothesis is either to block the action of gene E in some patches (so that those patches remain brown, and are not converted to white), or to reverse the effect of gene E in some patches (white is converted back to brown in those patches):
This hypothesis predicts that E should be epistatic to B -- i.e., in the absence of wild type gene E, the phenotype should be fully brown, regardless of whether gene B is wild type or mutant. This is exactly what we do see -- i.e., the phenotype of ee is the same that of bbee -- fully brown. Therefore, this hypothesis IS CONSISTENT with the data; one of the these pathways must be correct:
CORRECT PATHWAY:
This pathway is also consistent with the cross described: the F2 progeny from the F1 cross (DdEe x DdEe) will be:
- 9 D_E_ : 3 D_ee : 3 ddE_ : 1 ddee
- wildtype brown black black
giving a 9 wild type : 4 black : 3 brown ratio, which is exactly what is seen.