| 1. | (a) |
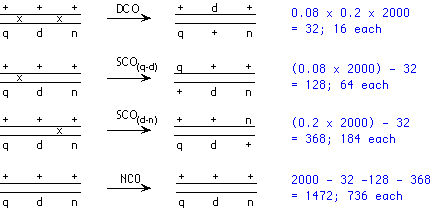
The F1 progeny are fully heterozygous, with the dominant alleles in cis (+++/+++ crossed with qdn/qdn giving +++/qdn). The testcross is +++/qdn x qdn/qdn. The progeny phenotypes and numbers are shown:
|
||||||||
| (b) |
Here, n is unlinked to q and d. So we can calculate the progeny numbers with respect to q and d; half of each of those classes will additionally be n+, and half of each class will be n. With respect to q and d, the crossover progeny will be + d and q + , and will account for (0.08 x 2000) = 160 progeny. Each of these two classes will be evenly split between n+ and n; likewise, each of the two non-crossover products (2000 - 160 = 1840 total) will also be split evenly between n+ and n:
|
|||||||||
| (c) |
Here, we can simply add up the progeny numbers from the two separate crosses and calculate the apparent map distances from those:
Note that if the assistant knew he was combining two separate maps, he could simply take the mean of the two (d-n) map distances and get the same answer.
|