HWP are based on following assumptions:
|
| Example Phenotype (codominant) | White | Pink | Red | |
| Example Phenotype (dominant) | White | White | Red | |
| Genotypes | AA | Aa | aa | |
| Observed numbers | NAA | NAa | Naa | |
| Genotype frequency | NAA/N | NAa/N | Naa/N | N=NAA+NAa+Naa |
| P | H | Q | P+H+Q=1 |
| Allele | A | a | |
| Allele frequency | pA [we use often just p] | pa [we use often just q] | pA+pa=1 |
| P + 0.5 H | Q + 0.5 H | ||
|
(2 NAA + NAa) ----------- 2 N |
(2 Naa + NAa) ----------- 2 N |
If the mentioned assumptions hold then the Hardy-Weinberg proportions are achieved in one generation, and after that the allele frequencies stay the same in all future generations.
| The observed numbers and the numbers expected from Hardy-Weinberg proportions for the MN blood group locus in an English sample of 1000 blood donors (from Cleghorn 1960) |
| Phenotype | Genotype | Observed Number | Expected Number |
| M | MM | 298 | p*p*T=294.3 |
| MN | MN | 489 | 2*p*q*T=496.3 |
| N | NN | 213 | q*q*T=209.3 |
| Total T | 1000 | 1000 |
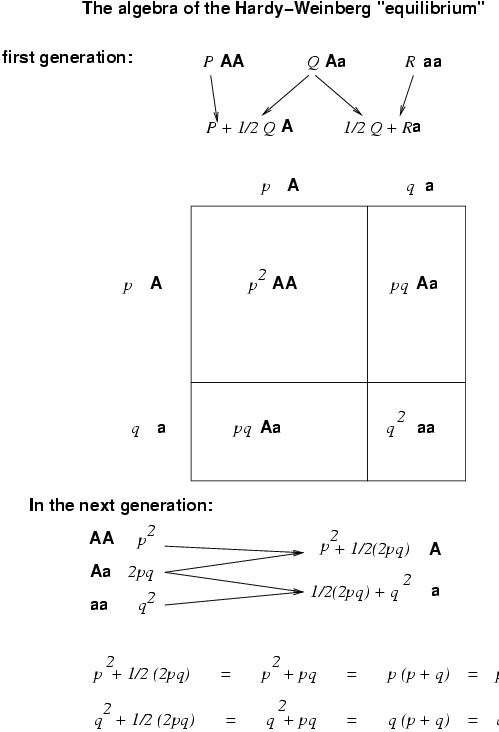
Use the "Algebra of the Hardy-Weinberg Equilibrium" to calculate the p and q of the MN blood locus example.
| If we assume that a population IS in Hardy-Weinberg proportions (HWP) then we can calculate the allele frequencies even in cases where one allele is dominant and we cannot distinguish between heterozygotes and the dominant homozygote. We use the fact that the frequency of the recessive allele (say a) has |
|
Paa = (frequency(a))2 = q2 q = frequency(a) = Sqrt(Paa) |
|
In Example 2 we calculate for the recessive
allele + in the life frogs is Paa = 834 / (834 + 53) = 0.940248 allele frequency q = paa = Sqrt(0.940248) = 0.969664 the dominant allele is p=1-q. |

 Two color morphs (alleles B and +) of the leopard frog (Rana pipiens). The allele B is dominant, heterozygotes (B+) look like BB. The study was done in Minnesota and investigated if there were differences in in HWP between animals found dead compared to animals caught alive. On the left the common subspecies (R. p. pipiens), on the right R. p. burnsi. Adapted from Hedrick (2000), data is from Merrell and Rodell (1968). Pictures are (c) Jeff LeClere. |
| Phenotype | Genotype | Observed Number | Allele Frequency | |
| Living | burnsi | BB, B+ | 53 | 0.030 |
| pipiens | ++ | 834 | 0.970 | |
| Dead | burnsi | BB,B+ | 39 | 0.018 |
| pipiens | ++ | 1050 | 0.982 |
| Example with 3 alleles | ||||||
| Genotypes | A1A1 | A1A2 | A1A3 | A2A2 | A2A3 | A3A3 |
| Genotype Frequencies | P11 | P12 | P13 | P22 | P23 | P33 |
| p(A1) = frequency(A1) = P11 + 0.5 P12 + 0.5 P13) |
| p(A2) = frequency(A2) = P22 + 0.5 P12 + 0.5 P23) |
| p(A3) = frequency(A3) = P33 + 0.5 P13 + 0.5 P23) |
| p(Ai) = frequency(Ai) = Pii + 0.5 Sumkj,i ≠ j(Pij) |
| The number of heterozygotes in a sample can be used as a measure of variability. The OBSERVED heterozygosity H is simply the frequency of the heterozygotes in the sample. If we assume HWP then the EXPECTED heterosygosity is 2pq for two alleles. For multiple alleles it is often easier to subtract the homozygote genotypes from 1, in general |
| Hexp= 1 - Sum(Pii) |
| In Example 2 the dominant allele is masking the heterozygotes, assuming that the population is in HWP, we can calculate the expected heterozygosity H = 2 * 0.969664 * 0.0303361 = 0.0588317 and by multiplying this with the total number (834+53) we estimate the number of heterozygotes as 52 (52.18). Most likely there is only one homozygote burnsi in the living frog sample. This is a common situation, rare alleles most often occur in a heteroyzgote. |
| We can test if a sample is in HWP by using a Chi2-Test with the Null hypothesis H0: Sample is in HWP, and the alternative hypothesis H1: Sample is NOT in HWP. For date sets with multiple allele one wants to use more sophisticated procedures, such as those by Guo and Thompson (1996). We use the following | |||||||||||||||||||||||||||||||||||||||
(Observed - Expected)2
Chi2 = Sum -------------------------------
Expected
|
|||||||||||||||||||||||||||||||||||||||
|
The sum is over all genotypic classes. From the calculated
Chi2 and with knowledge of another number, the
degrees of freedom, we can obtain by comparison with a tabulated
Chi2 the probability that the observed numbers deviates from
the expected numbers as much or more by chance. The degree of freedom
is the number of classes minus 1 and then minus the number of parameters
estimated from the data. For the two allele case
we have 3 genotypic classes (PAA,PAa,Paa)
of which one can be calculated by knowing the other two.
We have one parameter pA (we can calculate the other
as pa=1-pA). This results in 1 degree of
freedom (3-1 - 1). If we use a significance level of 5%
(we will erroneously accepting the alternative hypothesis in 5%
of all tested cases) the tabulated Chi2 is 3.84.
We will accept the alternative hypothesis when our testvalue
is bigger than the tabulated one.
In the two allele case we calculate |
|||||||||||||||||||||||||||||||||||||||
The follwing numbers are from Example 1, remark the use of observed and expeted numbers instead of frequencies.
|