








Questions about UW Pharmacology?
Here are a couple of our most popular questions about studying Pharmacology at UW.


Innovative, out-of-the box thinking has led to major breakthroughs in Pharmacology.
Our fresh approaches and groundbreaking science have made us vanguards in the field.

When people with different views exchange ideas, those people become a network. And networks test and refine those ideas, creating a proving ground for important discoveries, and powerful solutions to difficult problems.

A thriving and vibrant community in a breathtaking natural setting is what makes Seattle the perfect environment to learn, create and pursue your scientific interests.

The more diverse a group is, the broader the cultural and intellectual milieu from which its ideas spring, the more powerful and far-reaching those ideas tend to be. At UW we foster and encourage diversity as essential to scientific advancement.
Read all about what we're doing in the department

Congratulations to Yasemin Sancak on her promotion to Associate Professor with tenure.

Excellence and leadership were defining characteristics of University of Washington Neurobiologist William (Bill) A. Catterall who died at age 77 on February 28, 2024. Born and raised in Providence Rhode Island, Bill was an Ivy League scholar, graduating from Brown and Johns Hopkins Universities. He joined the Department of Pharmacology at the University of Washington School of Medicine in 1977 and was research-active until his death. His contributions to modern pharmacology, leadership impact at UW, and the legacy of his trainees are incalculable. He is survived by his wife Tina, son Douglas, and daughter Elizabeth.
David Shechner (UW) and Gene Yeo (UC, San Diego) received the Chan-Zuckerberg Collaborative Pairs Award. Their project looks to leverage and further develop new RNA-targeted microenvironment-mapping tools in patient-derived neurons,...

ASPET is pleased to award Dr. John D. Scott, PhD from the University of Washington, the 2024 Julius Axelrod Award in Pharmacology. The Axelrod Award was established in 1991 to honor the memory of the eminent American pharmacologist who shaped the fields of neuroscience, drug metabolism, and biochemistry and who served as a mentor for numerous eminent pharmacologists around the world.

Maryanne Kihiu awarded 2023 AHA Predoctoral Fellowship. The fellowship is meant to enhance the training of promising students in pre-doctoral or clinical health professional degree training programs and who intend...

Bill Catterall, Professor of Pharmacology at the University of Washington, will receive the 2023 Lifetime Achievement Award of the International Union for Basic & Clinical Pharmacology (IUPHAR) in recognition of...

Ion Channel Pharmacology & Genetics, Autumn Quarter, 2023 (2 credits) Course Chairs: Bill Catterall and Yasemin Sancak, Pharmacology Combined Lectures and Discussion Open to interested graduate students, postdoctoral fellows, and research...

On April 23, 2023, Akira (Aki) Horita, Professor Emeritus, died peacefully at his home at the age of 94. He was one of the earliest faculty members in the Department...
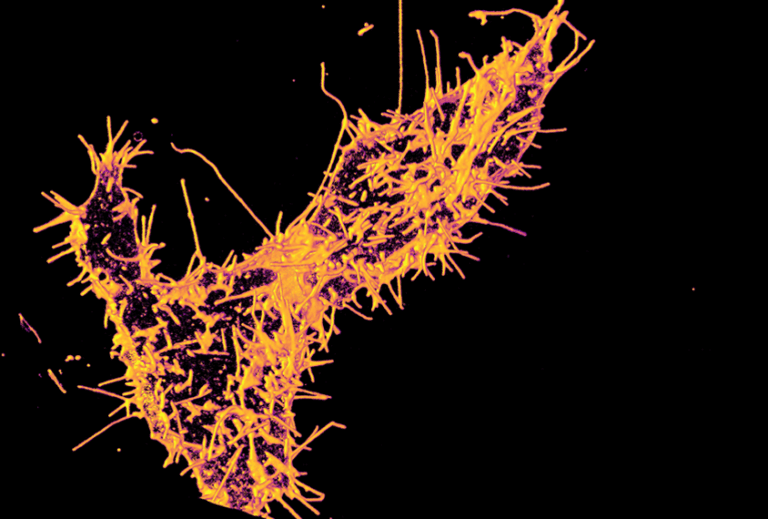
The Yadav lab’s newly published Science Signaling paper was featured in Spectrum. It is the go-to destination for the latest news and analysis about autism research.
A new paper from the Yadav lab was published in Science Signaling. Mutations in TAOK1, which encodes a serine-threonine kinase, are associated with both autism spectrum disorder (ASD) and neurodevelopmental delay...

UW Pharmacology is consistently ranked among the best in the nation and worldwide.




Here are a couple of our most popular questions about studying Pharmacology at UW.
GRE Scores are not required to apply to our graduate program.
We consider top applicants who have prior research experience that can add to the diversity of our program. If you wish to apply, visit our admissions section for more information. To get started on your application, apply to the Graduate School on-line at https://www.grad.washington.edu/applForAdmiss/
Please visit the UW Graduate School website for information on who qualifies for fee waivers, how to apply and FAQ’s at https://www.grad.washington.edu/admission/application-fee-waivers/
The current time to earning the PhD is 5.5 years. This of course depends on your field of study, and how long it takes to complete your dissertation. For a year by year breakdown of the degree requirements please visit this page.
If interested in a postoctoral fellowship, we encourage you to contact the faculty member you wish to study with, directly. Please go to our people page and click on primary faculty. To learn more about their research, browse the Research section.

YOUR PHARMACOLOGY ADVENTURE STARTS NOW
A degree from the University of Washington in Pharmacology can open up career opportunities in academics, industry, policy, and venture investing.
ADMISSIONS