
Multiplexed Genetic Engineering in Saccharomyces cerevisiae
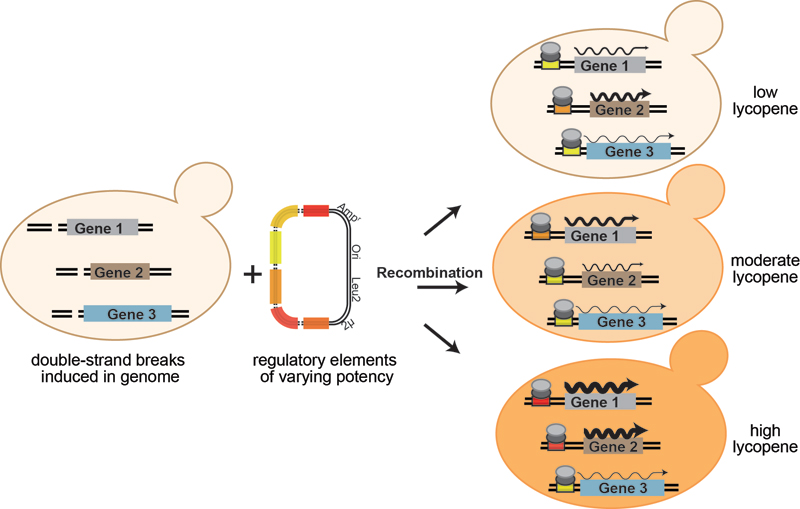
Strategies to optimize a metabolic pathway often require a large collection of strains to be generated, each containing different versions of the sequences that regulate the expression of the relevant pathway genes. Here we develop a set of reagents and methods to carry out this process at high efficiency in the yeast Saccharomyces cerevisiae. This toolkit includes a set of variants of the tet operator, which in conjunction with a TetR-VP16 activator drive expression over a 100-fold range; the induction of the I-OnuI homing endonuclease to target its recognition site in a gene to be modified, which boosts homologous recombination more than 105 over that in the absence of a double-strand break; and the generation of a plasmid carrying the six variant tet operator sites flanked by I-OnuI sites, uncoupling the transformation and recombination steps. As proof of principle, we introduce into the S. cerevisiae genome the three crt genes from Xanthophyllomyces dendrorhous required for yeast to synthesize lycopene, and carry out the recombination process to produce a population of cells with permutations of tetO variants regulating the three genes. We identify 0.7% of the cells as making lycopene, of which the vast majority have undergone a recombination event at each of the crt genes. Based on sequence analysis of these genes in strains that do not produce lycopene, we estimate a rate of ~20% recombination per targeted site, much higher than obtained in other studies.
Genetic Sensors for the Detection of Biosynthetic Products
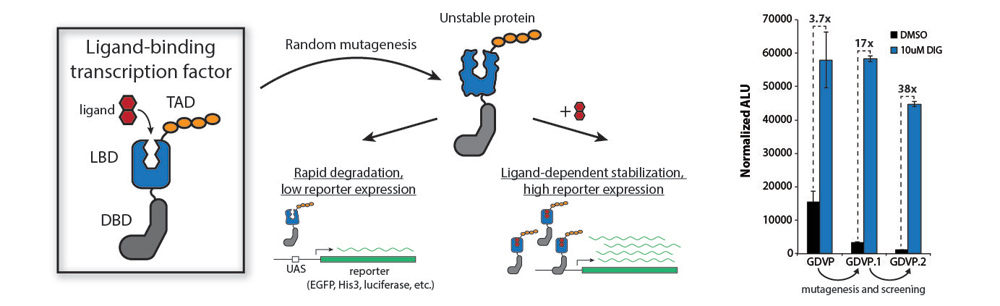
Model microorganisms such as E. coli and S. cerevisiae are valuable tools for the production of small molecules, such as therapeutics and biofuels, but the optimization of biosynthetic pathways in a new host is non-trivial. Identifying genetic modifications that enhance metabolite synthesis can be an exceptionally laborious process, particularly in the absence of a method to easily determine product yields. Genetically encoded biosensors that couple small molecule recognition to a readily measured output allow more rapid identification of cells with enhanced biosynthetic production or conditions that promote enhanced production. One of the most common mechanisms in nature for detecting the presence of a small molecule – be it a metabolite or an environmental agent – are biosensors that regulate the transcription of one or more genes upon binding of the relevant ligand. The goal of this project, carried out in collaboration with the labs of David Baker (University of Washington) and George Church (Harvard), is to design a transcription factor (TF)-based strategy for biosensors that may provide a general solution to the problem of small molecule detection.
Our strategy is to use destabilizing mutations that lead to degradation of the biosensor until it binds its cognate ligand. It should be possible to design a protein-based biosensor for any ligand as long as a ligand-binding domain (LBD) exists – or can be designed – for the desired small molecule. By fusing an unstable LBD to a TF, we couple ligand-dependent stabilization to reporter gene expression. As a proof of principle, we chose to use a de novo designed LBD, DIG10.3, which binds digoxigenin, a steroid similar to drugs used to treat heart failure. By fusing this protein to a DNA-binding domain (DBD) and a transcriptional activation domain (TAD), we were able to generate a ligand-dependent TF, which we designated GDVP. From a library of mutants, FACS analysis allowed us to identify destabilizing mutations that improved sensor function by >10-fold (GDVP.1 and GDVP.2).
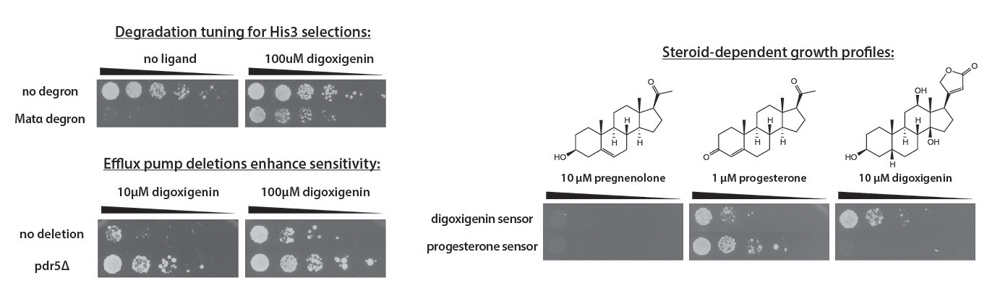
In order to tune the sensor for selections using a HIS3 reporter, we fused the degron from the Matα2 protein to GDVP to increase degradation. The fusion generated a sensor that leads to growth in histidine-deficient media more than 3 orders of magnitude better when the ligand (digoxigenin) is present. Sensitivity to digoxigenin can be further improved by deleting yeast efflux pumps, like Pdr5.
While the biosynthetic pathway required to produce digoxigenin is not known, other steroids such as progesterone have been successfully produced in yeast. An additional round of mutagenesis and screening allowed us to create a sensor that is extremely selective for progesterone but not its biosynthetic precursor, pregnenolone. Collaborators have used this sensor to identify mutations in 3-beta-hydroxysteroid dehydrogenase that improve progesterone bioproduction in yeast. We are currently applying these methods to develop sensors for new small molecules.
•Ben Jester
Engineering a biosensor for 3’-5’ Phosphoadenosine phosphate
3’-5’ Phosphoadenosine phosphate (pAp) is produced by removal of a sulfate group from 3’-phosphoadenosine 5’-phosphosulfate (PAPS), which is the most common coenzyme in the sulfotransferase reaction. pAp is converted into AMP and Pi by a pAp phosphatase (Met22). The pAp level in the cell is normally not detectable; however, pAp accumulates when Met22 activity is impaired. The accumulation of pAp is toxic to the tRNA quality control pathway by inhibiting a critical RNA processing enzyme activity.
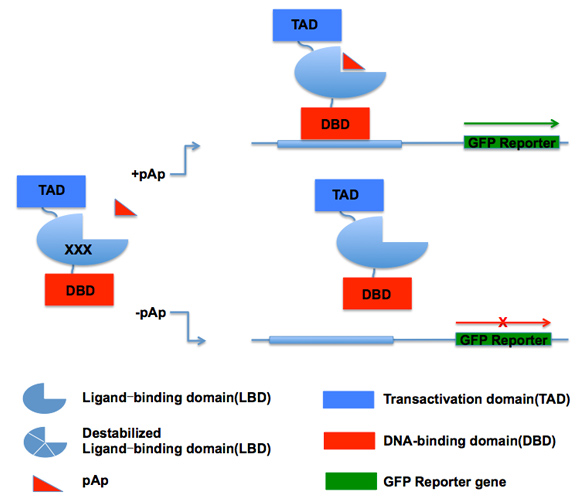
A biosensor for pAp concentration would both indicate whether the tRNA quality control machinery is active, and report on the methionine synthesis pathway. I am using protein engineering to create such a biosensor by generating a hybrid protein that contains a DNA-binding domain (DBD), a transcriptional activation domain (TAD) and a pAp ligand-binding domain (LBD). I will destabilize this protein via mutation, such that it will be degraded and not activate transcription. Binding of pAp will stabilize the hybrid protein and turn on expression of the reporter gene, GFP, thus reporting on the level of pAp.
I have selected part of the Met22 protein as a ligand-binding domain for pAp and confirmed that this domain has lost catalytic activity. I have mutagenized this domain to generate a library that is fused to a transcription activation domain and a DNA-binding domain. The library is transformed into a yeast strain with a GFP reporter gene that binds to the DBD. By sorting for fluorescence under conditions of either high pAp or low pAp, I should be able to identify an appropriate biosensor, and characterize the mutations present in it.
•Wei Zhou
Engineer a biosensor for detection of HPV16-L1 production
HPV human papillomaviruses (HPV) are common pathogens associated with a variety type of cancers, especially cervical cancer, which is the second most common cancer in women in United State. HPV16 infection is responsible for up to 50% of all cervical cancers.
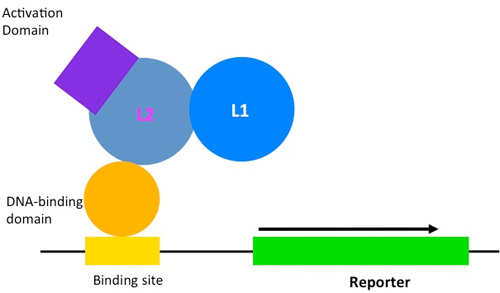
Successful preventive measures for HPV16 infection rely on vaccination. The vaccine is a virus-like particles generated from HPV16-L1 protein. When manufacturing the vaccine, viral capsid L1 gene is incorporated in yeast genome first and then the yeast produces HPV16 L1 capsid protein and it will be self-assembled to virus-like particles (vaccine). For many years, people are interested in using metabolic engineering the yeast genome to increase the production of L1 capsid protein. My project focus on engineering a biosensor for L1 capsid protein and using high-throughput way to screen those genome mutants which could increase L1 production.
My design strategy is to use destabilizing mutations that impair functional expression of the biosensor until it binds to L1. To do that, I tried HPV-16 L2 protein as the ligand-binding domain for L1. By fusing it with an activation domain and DNA-binding domain, we couple ligand-dependent stabilization to reporter gene expression.
|
        |
|
|
