|
Charge
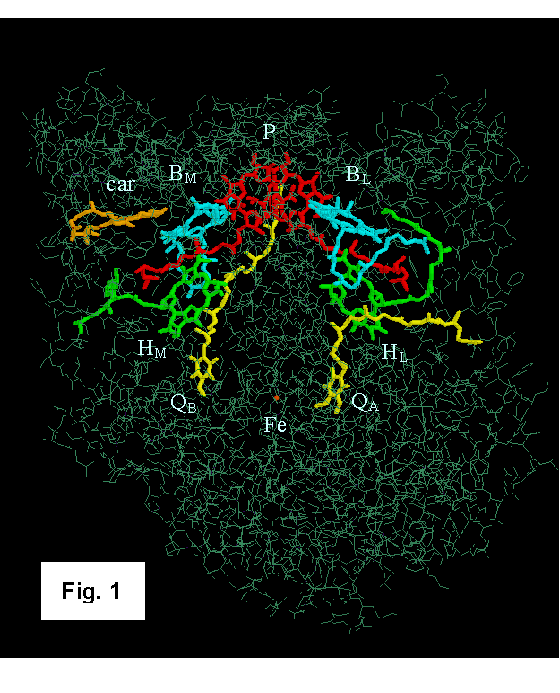
Separation in Photosynthetic Reaction Centers Reaction centers (RCs) are pigment-protein complexes that carry out the initial photochemical electron-transfer reactions of plants and photosynthetic bacteria. The structures of RCs from the purple bacterial species Rhodopseudomonas viridis and Rhodobacter sphaeroides have been solved by X-ray crystallography [1-8]. Figure 1 shows the Rb. sphaeroides structure as described by Ermler et al. [6]. The complex contains four molecules of bacteriochlorophyll (BChl), two molecules of bacteriopheophytin (BPh), two quinones and an iron atom, all bound to three polypeptides. Two of the BChls form a dimer that is often called the “special pair” (P). The other BChls (BL and BM), the BPhs (HL and HM), and the quinones (QA and QB) extend from P in two branches on either side of an axis of approximate symmetry. A 180˚ rotation about this axis interchanges the corresponding pigments on the two branches, along with the corresponding amino acid residues of the L and M subunits. The L and M subunits have very similar structures, and many of the corresponding residues either are identical or involve conservative replacements. However, a carotenoid (car) that plays mainly a protective role is located asymmetrically near BM. When RCs are excited with light, the
excitation energy moves to P within about 100 fs. The excited dimer
(P*) then transfers an electron to one of the BPhs (HL)
with a time constant of about 3 ps [9-17]. An electron
moves from the reduced BPh ( Reaction centers provide ideal systems for exploring relationships between protein structure and function. First, they carry out a variety of energy- and electron-transfer reactions ranging in time scale from 10-14 to 102 s. Because these reactions can be started with precise timing by a short flash of light, their kinetics can be measured over a broad range of temperatures or under other conditions such as in the presence of an oriented electric or magnetic field, and the electron carriers provide spectroscopic signatures that lend themselves to such measurements. The protein structures are known to high resolution and can be modified easily by site-directed mutagenesis. In addition, the initial reactions occur on the short time scales that are accessible to microscopic (all-atom) computer simulations. Although electron-transfer reactions between bound groups in proteins involve relatively simple chemistry, the factors that control their speed, specificity and temperature dependence are fundamentally the same as those that control other enzymatic processes. The following sections describe some of the recent experimental and computational studies of the initial electron-transfer steps in bacterial RCs, with an emphasis on work in our lab. The Initial Steps of Charge SeparationBecause
bacteriochlorophyll BL is situated between
P and HL (see Fig. 1), it seems reasonable to suggest that an
electron first jumps from P* to BL to form a However, this
scheme assumes that the energies of P*, Initial
attempts to detect As
mentioned above, the distinction between superexchange
and the two-step mechanism hinges largely on the energy of Figure
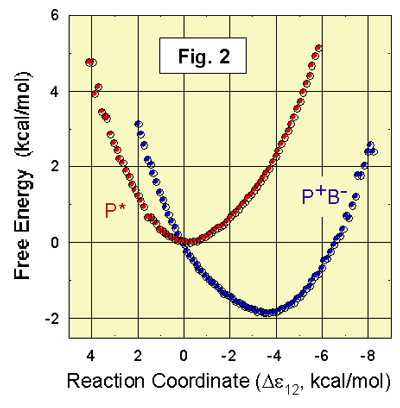
2 shows the results of recent calculations of the free-energies of P*BL
and If
the fluctuating, time-dependent energy gap is calculated over a sufficiently long
trajectory, the relative free energies of Here Dgi(x)
is the free energy of state i, Pi(x)
is the average probability of finding an energy difference De12 = x at any given time during a trajectory in state i, kB is
the Boltzmann constant, T is the temperature, and The kinetics of many electron-transfer reactions can be described well by the semiclassical Marcus equation [45,46]: In
this expression, k12 is
the rate constant, s12 is an
electronic coupling factor, If In
the simulations shown in Fig. 2, the calculated Experimental
estimates of the free energy of Since
The
calculated energy gap De12(t)
between P* and Similar
calculations put Before
such microscopic computer simulations of complex systems were possible, the
parameters s12 and l were obtained for many systems by fitting
the Marcus equation to experimental data on the rate constant as functions of Effects of Mutations on the Electron-Transfer Kinetics and SpecificityThe
effects of changing the free energy of Mutations
of residues that form hydrogen bonds to the BChls of
P also affect the charge-separation dynamics [15,65-69]. In wild-type RCs from Rb. sphaeroides, for example, the acetyl group of one of
the BChls of P forms an H-bond to H(L)168. Removing this bond lowers the midpoint
reduction potential (Em) of P, and thus
should stabilize In
addition to affecting the rate of electron transfer, mutations of Y(M210) and the homologous residue in the L polypeptide
(F(L181)) alter the specificity of charge separation. In recent work, Kirmaier
et al. [70] obtained about 30% “wrong way” electron transfer to HM
by combining the double mutation Y(M210)F/F(L181)Y with an additional mutation
that, by itself, had little effect on the specificity but facilitated the
experimental resolution of Low-frequency structural fluctuations of
polar groups like the phenolic OH group of tyrosine
M210 could explain the additional experimental observation that the
electron-transfer kinetics are multiphasic. Although the OH group of tyrosine M210
probably spends most of the time in an orientation that stabilizes
In recent work, we found that mutations of Arg (L135) or (M164) to Leu or Glu caused small shifts of the Em and absorption spectrum of P, but had very little effect on the charge-separation kinetics [74]. These residues occupy homologous positions on either side of the RC, with their ionizable groups about 14.5 Å from the center of P. Their electrostatic interactions with P clearly are very strongly screened. Although the ionizable groups of the Arg residues are almost completely buried in the protein, this screening could result largely from counterions in the nearby solvent, which keep the net charge of the system effectively constant. We have used the effects of these and other mutations on the Em of P to test computational methods for treating dielectric screening in proteins. Vibrational Modes, Wavepackets and RelaxationsThe initial electron-transfer steps from P* to Some
of the oscillations seen after excitation of reaction centers with short
flashes could reflect oscillations in the formation of Yakovlev et al. [81] suggest that P* and How
rapidly would where
the u(t) are the fluctuations of the energy gap about its mean value (u(t)
= De12(t) - The
frequencies of the vibrational modes that are coupled
strongly to electron transfer can be extracted by taking a Fourier transform of
the autocorrelation function A(t). Such a transform gives
the power spectrum of the fluctuations of De12(t). A particular mode will contribute to the
fluctuations of De12(t) if the vibrational
potential energy function is shifted along the vibrational
coordinate in The power
spectrum of De12(t) for P* ®
   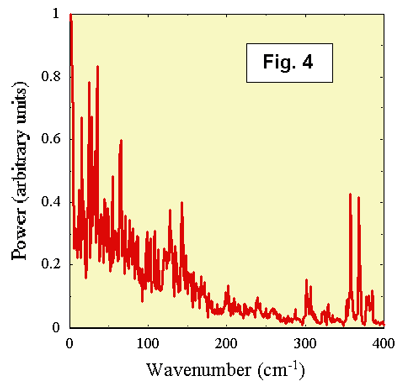 References1. Deisenhofer J, Epp O, Miki K,
Huber R & Michel H (1985) Structure
of the protein subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3 Å resolution. Nature 318:
618-624. 2. Deisenhofer J, Epp O, Sinning I
& Michel H (1995) Crystallographic
refinement at 2.3 Å resolution and refined model of the photosynthetic reaction
centre from Rhodopseudomonas viridis.
J. Mol. Biol. 246: 429-457. 3. Allen
JP, Feher G, Yeates TO,
Komiya H & Rees DC (1987) Structure
of the reaction center from Rhodobacter sphaeroides R-26: the protein subunits. Proc. Natl.
Acad. Sci. USA 84:
6162-6166. 4. Allen
JP, Feher G, Yeates TO,
Komiya H & Rees DC (1987) Structure
of the reaction center from Rhodobacter sphaeroides R-26: the cofactors. Proc. Natl. Acad. Sci. USA 84:
5730-5734. 5. Chang
CH, El-Kabbani O, Tiede D,
Norris J & Schiffer M (1991) Structure of the membrane-bound protein photosynthetic reaction center
from Rhodobacter sphaeroides.
Biochem.
30: 5352-5360. 6. Ermler U, Fritzsch G, Buchanan SK
& Michel H (1994) Structure of the
photosynthetic reaction centre from Rhodobacter sphaeroides at 2.65 Å resolution:
cofactors and protein-cofacter interactions.
Structure 2: 925-936. 7. McAuley KE, Fyfe PK, Ridge JP, cogdell
RJ, Isaacs NW et al. (2000) Ubiquinone binding, ubiquinone exclusion, and detailed cofactor conformation in
a mutant bacterial reaction center. Biochem. 39:
15032-15043. 8. Fyfe
PK & Jones MR (2000) Re-emerging
structures: continuing crystallography of the bacterial reaction centre. Biochim. Biophys. Acta Bioenerg. 1459:
413-421. 9. Holten D, Windsor MW, Parson WW & Thornber
JP (1978) Primary photochemical processes
in isolated reaction centers of Rhodopseudomonas viridis. Biochim. Biophys Acta
501: 112-126. 10. Woodbury
NW, Becker M, Middendorf D & Parson WW (1985) Picosecond kinetics of the initial photochemical
electron-transfer reaction in bacterial photosynthetic reaction center. Biochem. 24: 7516-7521. 11. Breton
J, Martin J-L, Migus A, Antonetti
A & Orszag A (1986) Femtosecond spectroscopy of excitation electron
transfer and initial charge separation in the reaction center of the
photosynthetic bacterium Rhodopseudomonas viridis. Proc. Natl. Acad. Sci.
USA 83: 5121-5175. 12. Holzapfel W, Finkele U, Kaiser W,
Oesterhelt D, Scheer H et
al. (1989) Observation of a bacteriochlorophyll anion radical during the primary charge
separation in a reaction center. Chem. Phys. Lett.
160: 1-7. 13. Holzapfel W, Finkele U, Kaiser W,
Oesterhelt D, Scheer H et
al. (1990) Initial electron transfer in
the reaction center from Rhodobacter sphaeroides. Proc. Natl. Acad. Sci.
USA 87: 5168-5172. 14. Kirmaier C & Holten D (1988) Subpicosecond spectroscopy of charge separation in Rhodobacter capsulatus reaction
centers. 15. Woodbury
NW, Peloquin JM, Alden RG, X. L, Taguchi AKW et al.
(1994) Relationship between
thermodynamics and mechanism during photoinduced
charge separation in reaction centers from Rhodobacter
sphaeroides. Biochem. 33: 8101-8112. 16. Martin
J-L, Breton J, Hoff AJ, Migus A & Antonetti A (1986) Femtosecond
spectroscopy of electron transfer in the reaction center of the photosynthetic
bacterium Rhodopseudomonas sphaeroides
R-26: direct electron transfer from the dimeric bacteriochlorophyll
primary donor to the bacteriopheophytin acceptor with
a time constant of 2.8 ± 0.2 psec. Proc. Natl.
Acad. Sci. USA 83:
957-961. 17. Paschenko VZ, Korvatovskii BN, Kononenko AA, Chamorovsky SK & Rubin AB (1985) Estimation of the rate of photchemical charge
separation in Rhodopseudomonas spheroides
reaction centers by fluorescence and absorption picosecond
spectroscopy. FEBS Lett. 191: 245-248. 18. Kaufmann
KJ, Petty KM, Dutton PL & Rentzepis PM (1975) Picosecond kinetics of events leading to reaction
center bacteriochlorophyll oxidation. Science 188: 1301-1304. 19. Rockley MG, Windsor MW, Cogdell
RJ & Parson WW (1975) Picosecond detection
of an intermediate in the photochemical reaction of bacterial photosynthesis.
Proc. Natl. Acad. Sci. USA 72: 2251-2255. 20. Wraight CA & Clayton RK (1973) The absolute quantum efficiency of bacteriochlorophyll
photooxidation in reaction centres
of Rhodopseudomonas spheroides.
Biochim.
Biophys Acta 333: 246-260. 21. Breton
J, Martin J-L, Fleming GR & Lambry J-C (1988) Low temperature femtosecond
spectroscopy of the initial step of electron transfer in reaction centers from
photosynthetic purple bacteria. Biochem. 27: 8276-8284. 22. Fleming
GR, Martin J-L & Breton J (1988) Rates
of primary electron transfer in photosynthetic reaction centers and their
mechanistic implications. Nature 33:
190-192. 23. Nagarajan
V, Parson WW, Davis D & Schenck CC (1993) Kinetics and free energy gaps of
electron-transfer reactions in Rhodobacter sphaeroides reaction centers. Biochem. 32: 12324-12336. 24. Kirmaier C, Holten D & Parson
WW (1985) Temperature and
detection-wavelength dependence of the picosecond
electron-transfer kinetics measured in Rhodopseudomonas
sphaeroides reaction centers. Resolution of new spectral
and kinetic components in the primary charge-separation process. Biochim. Biophys. Acta 810: 33-48. 25. Lauterwasser C, Finkele U, Scheer. H & Zinth W (1991) Temperature dependence of the primary
electron transfer in photosynthetic reaction centers from Rhodobacter
sphaeroides. Chem. Phys. Lett.
183: 471-477. 26. Kirmaier C, Holten D & Parson
WW (1985) Picosecond photodichroism
studies of the transient states in Rhodopseudomonas sphaeroides reaction centers at 5 K: effects of electron
transfer on the six bacteriochlorin pigments. Biochim. Biophys. Acta 810: 49-61. 27. Bylina EJ, Kirmaier C, McDowell L, Holten D
& Youvan DC (1988) Influence of an amino-acid residue on the optical properties and
electron transfer dynamics of a photosynthetic reaction centre complex. Nature 336: 182-184. 28. Kellog EC, Kolaczkowski S, Wasielewski MR & Tiede DM
(1989) Measurement of the extent of
electron transfer to the bacteriopheophytin in the
M-subunit in reaction centers of Rhodopseudomonas viridis. Photosynth. Res. 22: 47-59. 29. Holzwarth AR & Muller MG (1996) Energetics and kinetics of radical pairs in reaction centers from Rhodobacter sphaeroides. A femtosecond
transient absorption study. Biochem. 35:
11820-11831. 30. Kennis JTM, Shkuropatov AY, van Stokkum IHM, Gast P, Hoff AJ et al. (1997) Formation of a long-lived P+BA- state
in plant pheophytin-exchanged reaction centers of Rhodobacter sphaeroides R26 at
low temperature. Biochem. 36:
16231-16238. 31. van
Stokkum IHM, Beekman LMP,
Jones MR, van Brederode ME & van Grondelle R (1997) Primary
electron transfer kinetics in membrane-bound Rhodobacter
sphaeroides reaction centers: a global and target
analysis. Biochem. 36:
11360-11368. 32. Creighton
S, Hwang J-K, Warshel A, Parson WW & Norris J
(1988) Simulating the dynamics of the
primary charge separation process in bacterial photosynthesis. Biochem. 27: 774-781. 33. Parson
WW, Chu ZT & Warshel A (1990) Electrostatic
control of charge separation in bacterial photosynthesis. Biochim. Biophys. Acta 1017:
251-272. 34. Alden
RG, Parson WW, 35. Alden
RG, Parson WW, Chu ZT & Warshel
A, Macroscopic and microscopic estimates
of the energetics of charge separation in bacterial
reaction centers, in Reaction Centers
of Photosynthetic Bacteria: Structure
and Dynamics, 36. Warshel A, Chu ZT & Parson WW
(1989) Dispersed polaron
simulations of electron transfer in photosynthetic reaction centers.
Science 246: 112-116. 37. Warshel A & Parson WW (1991) Computer simulations of electron transfer
reactions in solution and photosynthetic reaction centers. Ann. Rev. Phys.
Chem. 42: 279-309. 38. Parson
WW & Warshel A, Simulations of electron transfer in bacterial reaction centers, in The Photosynthetic Reaction Center,
Norris, J and Deisenhofer, J, Editors. 1993, Academic
Press: 39. Parson
WW & Warshel A, Theoretical analyses of electron-transfer reactions, in Anoxygenic Photosynthetic Bacteria, Blankenship,
RE, Madigan, MT, and Bauer, CE, Editors. 1994, Academic
Publishers. 559-575. 40. Warshel A, Chu ZT & Parson WW
(1994) On the energetics of
the primary electron-transfer process in bacterial reaction centers. J. Photochem. Photobiol. A: Chem. 82:
123-128. 41. Warshel A, Chu ZT & Parson WW
(1997) Two-dimensional free energy
surfaces for primary electron transfer in a photosynthetic reaction center.
Chem. Phys. Lett. 265: 293-296. 42. Parson
WW, Chu ZT & Warshel A (1998) Reorganization
energy of the initial electron-transfer step in photosynthetic bacterial
reaction centers. Biophys. J. 74: 182-191. 43. Parson
WW, 44. Warshel A &
Parson WW (2001) Dynamics of biochemical and biophysical
reactions: insight from computer simulations. Quart.
Rev. Biophys. 34:
563-679. 45. Marcus
RA (1956) On the theory of oxidation-reduction reactions
involving electron-transfer. 46. Marcus
RA & Sutin N (1985) Electron transfers in chemistry and biology. Biochem. Biophys. Acta
811: 265-322. 47. Parson
WW, Chu ZT & Warshel A,
Free energy functions for charge
separation in wild-type and mutant bacterial reaction centers, in Photosynthesis: Mechanisms and Effects, Garab, G, Editor. 1998, Kluwer Acad. Publ.: Dordecht. 703-706. 48. Meade
TJ, Gray HB & Winkler JR (1989) Driving-force
effects on the rate of long-range electron transfer in ruthenium-modified cytochrome-c. J. Am. Chem. Soc. 111: 4353-4356. 49. Schmidt
S, Arlt T,
Hamm P, Huber H, Nägele T et al. (1995) Primary electron-transfer dynamics in
modified bacterial reaction centers containing pheophytin-a
instead of bacteriopheophytin-a. Spectrochim.
Acta A 51A: 1565-1578. 50. Shkuropatov AY & Shuvalov VA
(1993) Electron transfer
in pheophytin a-modified reaction centers from Rhodobacter sphaeroides (R-26).
FEBS. Lett. 322: 168-172. 51. Arlt T, Dohse B, Schmidt S, Wachtveitl J, Laussermaier E et
al. (1996) Electron transfer dynamics of Rhodopseudomonas viridis reaction
centers with a modified binding site for the accessory bacteriochlorophyll.
Biochem.
35: 9235-9244. 52. Lin
S, Taguchi AKW & Woodbury NW (1996) Excitation
wavelength dependence of energy transfer and charge separation in reaction
centers from Rhodobacter sphaeroides:
Evidence for adiabatic electron transfer. J. Phys. Chem. 100: 17067-17078. 53. van
Brederode ME, Jones MR, van Mourik
F, van Stokkum IHM & van Grondelle
R (1997) A new pathway for transmembrane
electron transfer in photosynthetic reaction centers of Rhodobacter
sphaeroides not involving the excited special pair.
Biochem.
36: 6855-6861. 54. van
Brederode ME, van Mourik F,
van Stokkum IHM, Jones MR & van Grondelle R (1999) Multiple pathways for ultrafast transduction of light energy in the
photosynthetic reaction center of Rhodobacter sphaeroides. Proc. Natl. Acad. Sci.
USA 96: 2054-2059. 55. Alden
RG, Parson WW, Chu ZT & Warshel
A (1996) Orientation of the OH dipole of
tyrosine (M)210 and its effect on electrostatic energies
in photosynthetic bacterial reaction centers. J. Phys. Chem. 100: 16761-16770. 56. Parson
WW, Nagarajan V, Gaul D, Schenck CC, Chu ZT et al., Electrostatic
effects on the speed and directionality of electron transfer in bacterial
reaction centers: the special role of tyrosine M-208, in Reaction Centers of Photosynthetic Bacteria,
Michel-Beyerle, M-E, Editor. 1990, Springer-Verlag: 57. Chan CK, Chen LXQ, Dimagno
TJ, Hanson DK, Nance SL et al. (1991) Mechanism
of the initial charge separation in photosynthetic bacterial reaction centers. Chem. Phys. Lett.
176: 366-372. 58. Finkele U, Lauterwasser C, Zinth W, Gray KA & Oesterhelt
D (1990) Role of tyrosine M210 in the
initial charge separation of reaction centers of Rhodobacter
sphaeroides. Biochem. 29: 8517-8521. 59. Gray
KA, Wachtveitl J & Oesterhelt
D (1992) Photochemical trapping of a bacteriopheophytin anion in site-specific reaction-center
mutants from the photosynthetic bacterium Rhodobacter
sphaeroides. Eur. J. Biochem. 207:
723-731. 60. Nagarajan
V, Parson WW, Gaul D & Schenck C (1990) Effect of directed mutations of the tyrosine
at site (M)210 on the primary photosynthetic electron
transfer process in Rhodobacter sphaeroides.
Proc. Natl. Acad. Sci. USA 87: 7888-7892. 61. Jia Y, DiMagno TJ, Chan C-K, Wang
Z, Du M et al. (1993) Primary charge separation in mutant reaction centers of Rhodobacter capsulatus. J.
Phys. Chem. 97: 13180-13191. 62. Shochat S, Arlt T, Francke C, Gast P, van Noort PI et al. (1994) Spectroscopic
characterization of reaction centers of the (M)Y210W mutant of the
photosynthetic bacterium Rhodobacter sphaeroides. Photosynth. Res. 40: 55-66. 63. Beekman LMP, van Stokkum IHM, Monshouwer R, Rijnders AJ, McGlynn P et al. (1996) Primary
electron transfer in membrane-bound reaction centers with mutations at the M210
position. J. Phys. Chem. 100:
7256-7268. 64. Parson
WW & Warshel A. Time-dependent free energy functions for charge separation in
photosynthetic bacterial reaction centers. in Abstracts, 39th Sanibel Symposium. 1999.
65. Williams JC, Alden RG, Murchison HA, Peloquin JM, Woodbury NW et al. (1992) Effects of mutations near the bacteriochlorophylls
in reaction centers from Rhodobacter sphaeroides. Biochem. 31:
11029-11037. 66. Allen
JP, Williams JC, Graige MS, Paddock ML, Labahn A et al. (1998) Free
energy dependence of the direct charge recombination from the primary and
secondary quinones in reaction centers from Rhodobacter sphaeroides. Photosyn.
Res. 55: 227-233. 67. Lin
X, Murchison HA, Nagarajan V, Parson WW, Allen JP et al. (1994) Specific alteration of the oxidation
potential of the electron donor in reaction centers from Rhodobacter
sphaeroides. Proc. Natl. Acad. Sci. USA 91:
10265-10270. 68. Mattioli TA, Williams JC, Allen JP & Robert B (1994) Changes in primary donor hydrogen bonding
interactions in mutant reaction centers from Rhodobacter
sphaeroides: Identification of the vibrational frequencies of all the conjugated carbonyl
groups. Biochem. 33: 1636-1643. 69. Arlt T, Bibikova M, Penzkofer H, Oesterhelt D & Zinth W (1996) Strong
acceleration of primary photosynthetic electron transfer in a mutated reaction
center of Rhodopseudomonas viridis.
J. Phys. Chem. 100: 12060-12065. 70. Kirmaier C, He C & Holten D
(2001) Manipulating the direction of
electron transfer in the bacterial reaction center by swapping Phe for Tyr near BChlM (L181) and Tyr
for Phe near BChlL
(M208). Biochem. 40:
12132-12139. 71. Zhang
LY & Friesner RA (1998) Ab initio calculation of electronic coupling in
the photosynthetic reaction center. Proc. Natl. Acad. Sci.
USA 95: 13603-13605. 72. Ivashin N, Källenbring B, Larsson
S & Hansson Ö (1998) Charge separation in photosynthetic reaction center. J. Phys. Chem.
B. 102: 5017-5022. 73. Kolbasov D & Scherz A (2000) Asymmetric electron transfer in reaction
centers of purple bacteria strongly depends on different electron matrix
elements in the active and inactive branches. J. Phys. Chem. B 104: 1802 -1809. 74. Johnson
E & Parson WW (2002) Electrostatic
interactions in an integral membrane protein. Biochem. submitted. 75. Vos MH, Rappaport F, Lambry J-H, Breton J & Martin J-L (1993) Visualization of coherent nuclear motion in
a membrane protein by femtosecond spectroscopy.
Nature 363: 320-325. 76. Vos MH, Jones MR, Hunter CN, J. B & Martin J-L (1994) Coherent nuclear dynamics at room
temperature in bacterial reaction centers. Proc. Natl. Acad. Sci. USA 91:
12701-12705. 77. Vos MH, Jones MR, Breton J, Lambry
JC & Martin J-L (1996) Vibrational dephasing of long- and short-lived primary donor excited
states in mutant reaction centers of Rhodobacter sphaeroides. Biochem. 35: 2687-2692. 78. Stanley
RJ & Boxer SG (1995) Oscillations in
spontaneous fluorescence from photosynthetic reaction centers. J. Phys.
Chem. 99: 859-863. 79. Streltsov AM, Aartsma TJ, Hoff AJ
& Shuvalov VA (1997) Oscillations within the B-L absorption band of Rhodobacter
sphaeroides reaction centers upon 30 femtosecond excitation at 865 nm. Chem. Phys. Letts. 266:
347-352. 80. Streltsov AM, Vulto SIE, Shkuropatov AY, Hoff AJ, Aartsma
TJ et al. (1998) BA and BB absorbance perturbations induced by coherent
nuclear motions in reaction centers from Rhodobacter sphaeroides upon 30-fs excitation of the primary donor.
J. Phys. Chem. B. 102: 7293-7298. 81. Yakovlev AG, Shkuropatov AC &
Shuvalov AV (2000) Nuclear wavepacket motion producing a reversible
charge separation in bacterial reaction centers. FEBS Lett. 466:
209-212. 82. Vos MH, Rischel C, Jones MR &
Martin J-L (2000) Electrochromic detection of a coherent component in the
formation of the charge pair P+HL- in
bacterial reaction centers. Biochem. 39: 8353 -8361. 83. Spörlein S, Zinth W & Wachtveitl J (1998) Vibrational coherence
in photosynthetic reaction centers observed in the bacteriochlorophyll
anion band. J. Phys. Chem. B 102:
7492-7496. 84. Yakovlev AG & Shuvalov VA
(2000) Formation of bacteriochlorophyll
anion band at 1020 nm produced by nuclear wavepacket
motion in bacterial reaction centers. J. Chin. Chem. Soc. 47: 1-6. 85. Kitzing E & Kuhn H (1990) Primary electron transfer in photosynthetic reaction centers. J.
Phys. Chem. 94: 1699-1702. 86. Peloquin JM, Williams JC, Lin X, Alden RG, Taguchi AKW et
al. (1994) Time-dependent thermodynamics
during early electron transfer in reaction centers from Rhodobacter
sphaeroides. Biochem. 33: 8089-8100. 87. Schulten K & Tesch M (1991) Coupling of protein motion to electron transfer: molecular dynamics and stochastic quantum
mechanics study of photosynthetic reaction centers. Chem. Phys. 158: 421-446. 88. Treutlein H, Schulten K, Brunger AT, Karplus M, Deisenhofer J et al. (1992) Chromophore-protein interactions and the function of the photosynthetic reaction
center: a molecular dynamics study. Proc. Natl. Acad. Sci.
USA 89: 75-79. 89. Marchi M, Gehlen JN, Chandler D
& Newton M (1993) Diabatic surfaces and the pathway for primary
electron transfer in a photosynthetic reaction center. J. Am. Chem. Soc. 115: 4178-4190. 90. Souaille M & Marchi M (1997) Nuclear dynamics and electronic transition in a
photosynthetic reaction center. J. Am. Chem. Soc. 119: 3948-3958. 91. Czarnecki K, Chynwat V, Erickson
JP, Frank HA & Bocian DF (1997) Characterization of the strongly coupled,
low-frequency vibrational modes of the special pair
of photosynthetic reaction centers via isotopic labeling of the cofactors.
J. Am. Chem. Soc. 119: 415-426. |