Tetrad Dissection
C. Tachibana, Young lab
April 2006
Background:
The yeast we work with (Saccharomyces
cerevisiae) will grow mitotically
as either a haploid or a diploid.
Mitotic growth produces clonal (genetically identical) cultures or
colonies. Two haploid cells will
mate, or fuse into a diploid cell, if they are of opposite mating types, called
a and a.
The resulting a/a diploid is induced to
sporulate, or undergo meiosis, by poor nutrient conditions. Like mammalian meiosis, the 4 cells
that result are genetically non-identical and haploid. Unlike mammalian meiosis, the 4 cells
that result from one cell going through meiosis stay together in a sac called
an ascus. The 4 cells together are
called a tetrad.
Sometimes we use mating and
sporulation to generate a new strain with a desired combination of gene
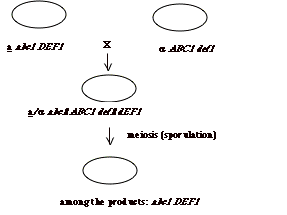
alleles. For example, if we have a
cells with a mutation in gene abc1
(yeast gene names have three letters, wildtype alleles are capitalized and
mutant are lowercase) and a cells with a mutation in
genes def1 and want a strain
that is genotype abc1 def1, we
can mate the two strains, induce meiosis in the resulting diploid and choose
the haploid spores that have the correct genotype.
Sometimes we use mating and
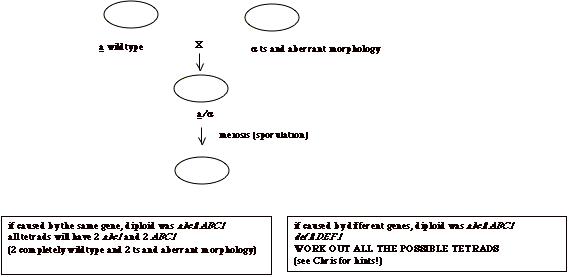
sporulation to detect gene linkage, for example to see if two phenotypes are
caused by the same, or by different genes. For example, if we have a strain with two phenotypes:
temperature sensitivity (ts) and aberrant cell morphology, and we want to see
if both phenotypes are caused by the same gene, we can cross the mutant strain
to a wildtype and sporulate the resulting diploid. If both phenotypes are caused by mutations in the same gene,
the ts and aberrant morphology will always segregate together. If they are caused by different,
unlinked genes, these will independently assort in meiosis and some spores will
have one phenotype but not the other.
Technique (always use sterile technique)
1. Sporulating:
a) Put 3-5 mls of pre-spor
medium in a tall white-capped tube.
b) Put in a small amount of diploid cells (this much: • )
c) Shake at room
temperature overnight.
d) The next day, spin 5
minutes at setting 2 or 2.5 in the swinging bucket centrifuge.
e) Remove pre-spor medium
and discard.
f) To the cell pellet add
3-5 mls of sporulation medium.
(They should not be very dense.
If they look like skim milk or denser, diltute it by taking 0.5 ml of
the cells and adding it to a new tube with 5 mls of sporulation medium).
g) Shake at room temperature
3-7 days.
h) Check every day by
removing about 10µl and putting on a glass slide. Cover with a cover slip and check under the microscope. At least 20% should look like tetrads
-- 4 cells clustered within a sac, before you start to dissect.
i) Put some YPD plates at
room temperature for a few days to dry out.
OR:
a) Patch thinly onto
sporulation medium plates (KAc).
Leave at 30 or room temp 3-14 days, checking every day as in step h)
above.
b) For difficult to
sporulate strains, thinly patch on pre-spor plates. The next day, transfer to sporulation plates by replica
plating or taking a small amount from pre-spor and patching onto spor.
2. Dissecting:
a) Take 0.1 - 0.5 mls
(100-500 µl) of cells from the sporulation medium and put it in a sterile
Eppendorf tube. If they are not
dense and you can see a lot of light through the culture, take 0.5 mls. If they are approaching skim milk, take
0.1 or less.
OR
a) Take about this much• from the sporulation plate and put in 500µl sterile
water in an Eppendorf tube.
b) Spin in the Eppendorf
centrifuge (microfuge) a few seconds.
c) Remove the liquid
supernatant and discard.
d) The cell pellet should
be barely visible (like this: • ) If it is bigger, resuspend in 1 ml water, remove 0.1 ml,
transfer to a new Eppendorf tube, spin for a few second, remove and discard the
supernatant and check the pellet again until it barely visible.
e) Wash once in water: add
1 ml sterile water. Pipet up and
down carefully to resuspend the cell pellet. Spin a few seconds in the Eppendorf centrifuge.
f) Remove the liquid
supernatant and discard.
g) Resuspend the cell
pellet in 20µl of zymolyase solution (0.5 mg/ml in 1M sorbitol, store at -20).
h) Incubate 8-10 minutes at
30o C
i) Slowly drop in 0.2 ml
sterile water and place the tube on ice.
j) Get a YPD plate that has
been drying at room temperature a few days. Use a permanent marker to make lines down the middle on the
bottom (agar containing) part. Make
one line 4 cm from one end and another 4.5 cm, so you have a narrow lane drawn
in the middle of the plate.
k) Take 10-15 µl of the
zymolyase-treated tetrads and drop it on the agar, at one end of the lane/
l) Tilt the plate so the
drop runs evenly down the lane to the other end. Let soak in/dry.
AT THE MICROSCOPE: ALWAYS WATCH OUT FOR THE DELICATE NEEDLE WHEN MOVING THE LENS, PLATFORM OR PLATE
m) Remove the cover and
petri plate lid from the Zeiss dissecting microscope. Carefully wipe around the plate holder and needle with
ethanol.
n) Turn on the light. Put the stick that moves the needle in
the middle of its range. Look in
the microscope to see if the needle is in the middle of the field of view. If not, move the entire needle holder,
very carefully, to get it in the middle.
For these initial adjustments, use the black 5X lens.
o) Adjust the platform so
the needle will be at the top and in the middle of the line of cells once you
put it on the scope, on the plate holder.
Remove the lid from the plate with the tetrads and invert it onto the
plate holder, with the line of cells oriented vertically.
p) From the side, look at
the needle, and dial the needle
holder up or down so it is close to, but not touching the agar surface.
q) Look through the
microscope and find the needle, moving the lens up and down carefully to find
it. Be sure not to move it so far
that the lens smashes into the plate.
If you have trouble finding it, carefully move the needle up toward the
agar by moving the stick on the left.
Gently stick the need in the agar to make a small hole, then adjust the
platform until the hole is in focus.
r) Adjust the needle up or
down so it can touch the agar and can move from side to side easily.
s) Move the platform with
the plate holder over to the line of cells and get them in focus. Switch to the 10X lens.
t) Start by cleaning the
needle. Gently pull the lever up
and embed the needle in the agar where there are no cells a few times. Do this anytime you are unsure if there
are cells on the needle and want to get them off.
u) Find a tetrad. It will look like 4 very small cells
held together in a diamond shape or a pile of tiny cannonballs with 3 on the
bottom and 1 on top. Make sure
they are well away from other cells or they might be 4 unrelated cells stuck
together.
v) Pick up the tetrad by
touching it with the needle. If
you have to, touch it several times.
If the cells come apart, try to pick them all up.
w) Once you have the 4 cells on the
needle, make sure it is clear of the agar. Move the platform to coordinates in one corner of the plate,
far enough from the stripe of cells that they will not grow over it (e.g. 5
(side) x 125 (top). Touch the
needle to the agar to remove the cells.
Use the needle to place cells into a line along one coordinate, leaving
one cell at each coordinate (e.g. 5 x 125, 5 x 130, etc).
x) Return to the line of
cells in the middle, pick up another tetrad and distribute the 4 cells in a
line next to the first line (e.g. 10 x
125, 10 x 130, etc). Continue
until the plate has as many tetrads as you can fit.
y) When finished, carefully
remove the plate and cover with its original lid. Incubate at 30oC overnight.
z) The next day, take a
ethanol-flame sterilized metal spatula and cut out and discard the line of
cells in the middle, before it overgrows the tetrads.
HINTS
• If tetrads won't
separate, put the tip of the needle just on the agar surface just touching the
tetrad. Gently bang on the table to shake the cells apart. You can also do this if cells won't
come off the needle. Beware of
making a big hole in the agar and burying the cells in it. Beware of bubbles this cause on the
agar surface that look like cells.
Bubbles disappear in a few seconds. Cells don't.
• If the plates are uneven,
watch out for the needle clearing the agar in one area and being too close to
the agar in another area.
• You can use the needle to
sweep away cells to make room to manipulate around a tetrad. Clean the needle afterwards by sticking
it into the agar a few times.

For more needles: www.corastyles.com