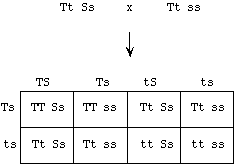
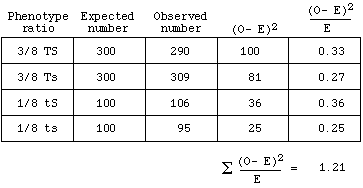
| 3. |
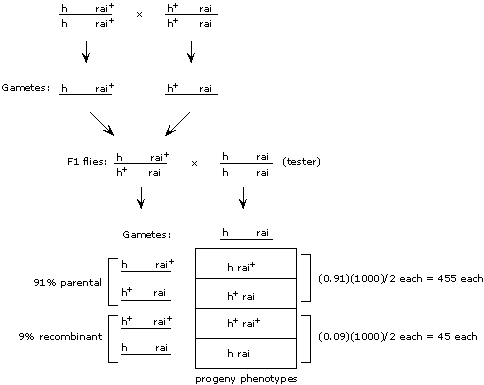
Remember that map distance = percent recombinant meiotic products = percent recombinant progeny.
To get the map distance, we need to know what fraction of the gametes from the heterozygous parent are recombinant. From (2) above, we already know the parental types for each cross except cross (ii); the other two in each case must be the recombinant. Then, for each cross, we can simply calculate what percent of all progeny are recombinant, and that would be the map distance between the two genes.
| Cross |
Recombinant progeny phenotypes |
Total # of recombinants |
Total # of progeny |
% recombination (= map distance) |
| i |
se+ h & se h+ |
5 + 4 = 9 |
821 |
(9/821)*100=1.1 |
| ii |
<can't tell> |
|
|
(50--assorting independently) |
| iii |
b+ rd+ & b rd |
12 + 15 = 27 |
862 |
(27/862)*100=3.1 |
| iv |
corr+ b & corr b+ |
44 + 51 = 95 |
835 |
(95/835)*100=11.4 |
| v |
corr+ rd+ & corr rd |
70 + 80 = 150 |
1009 |
(150/1009)*100=14.9 |
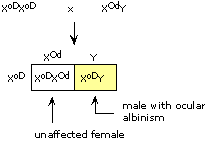
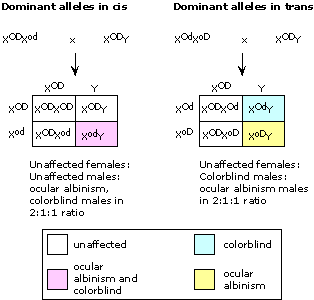
Thus, the se locus and the h locus are linked, and b, rd, and corr loci are linked. se assorts independently of b. Because h is tightly linked to se, h will probably assort independently of se also. Putting all the data together, se and h form a linkage group, as do b, rd, and corr. It is not possible to tell from the data whether se and h are truly unlinked from b, rd, and corr (i.e., located on separate chromosomes), or whether the two groups of genes are 50 or more map units apart on the same chromosome. The completed maps (not to scale!) are:
se----1.1 cM ---h
|---------------|
-
-
- rd-----3.1 cM-----b--------- -11.4 cM------------corr
- |-----------------|-------------------------------|
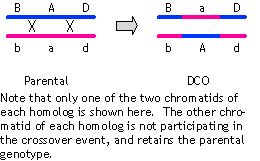
The two intervals in the bottom map don't quite add up to the 14.9 cM seen from the rd-corr cross; in this instance, because the data don't allow any other order of genes, we must conclude that simplest explanation for the slight discrepancy is just experimental error.
(Note that there is not a separate map for the '+' alleles; a map drawn this way shows the loci, not the alleles--so 'rd' in this map refers to the position of the rd locus on the chromosome relative to the other loci.)
|
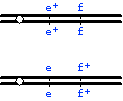 or
or