|
The Odom Laboratory
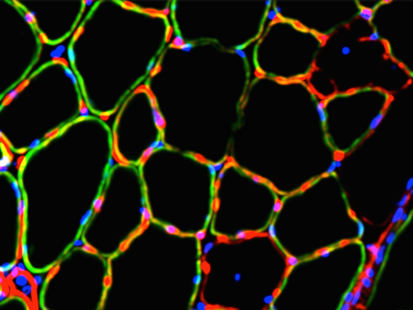 Dr. Guy Odom is a Research Assistant Professor in the Department of Neurology at the University of Washington; whose research focuses on neuromuscular disease with an emphasis on the muscular dystrophies (MD’s). As an example, gene mutations that affect production of the protein dystrophin are the cause of Duchenne muscular dystrophy (DMD) the most common lethally inherited disorder of children. A primary research interest in the Odom lab is to develop a more thorough understanding of the inherent muscle biology occurring during MD disease progression. Additionally, to identify contributory protein-protein interactions, biomarkers, & the like as potential therapeutic targets. As such, the labs’ research generally involves developing or improving genetic-based therapies. To that end, approaches often utilized include viral vector technology, high-resolution microscopy, omics technologies, incorporation of machine learning-based modeling, applying non-invasive imaging techniques & evaluating striated muscle contractile performance in dystrophic animal models. Dr. Guy Odom is a Research Assistant Professor in the Department of Neurology at the University of Washington; whose research focuses on neuromuscular disease with an emphasis on the muscular dystrophies (MD’s). As an example, gene mutations that affect production of the protein dystrophin are the cause of Duchenne muscular dystrophy (DMD) the most common lethally inherited disorder of children. A primary research interest in the Odom lab is to develop a more thorough understanding of the inherent muscle biology occurring during MD disease progression. Additionally, to identify contributory protein-protein interactions, biomarkers, & the like as potential therapeutic targets. As such, the labs’ research generally involves developing or improving genetic-based therapies. To that end, approaches often utilized include viral vector technology, high-resolution microscopy, omics technologies, incorporation of machine learning-based modeling, applying non-invasive imaging techniques & evaluating striated muscle contractile performance in dystrophic animal models.
Publications
Selected Peer-Reviewed Publications
Listed here are the most relevant to our current research interests.
- Odom GL, Gregorevic P, Allen JM, Finn EE, Chamberlain JS. Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol Ther. 2008 Sep;16(9):1539-45. (PMID: 18665159)
- Odom GL, Banks GB, Schultz BR, Gregorevic P, Chamberlain JS. Preclinical studies for gene therapy of Duchenne muscular dystrophy. J. Child Neurol. 2010 Sep; 25(9):1149-57. (PMID: 20498332)
- Odom GL, Gregorevic P, Allen JM, Chamberlain JS. Gene therapy of mdx mice with large truncated dystrophins generated by recombination using rAAV6. Mol Ther. 2011;19(1): 36-45. (PMID: 20859263)
- Bieber S, Halldorson JB, Finn E, Chamberlain JS, and Odom GL. Extracorporeal Delivery of rAAV with Metabolic Exchange and Oxygenation. Nature Scientific Reports. 2013;3:1538. (PMID: 23528884)
- Banks GB, Combs A, Odom GL, and Chamberlain JS. Muscle structure influences utrophin expression in mdx mice. PLOS Genetics, 2014; Jun 12;10(6). (PMID: 24922526)
- Arnett A, Konieczny P, Ramos JN, Hall J, Odom GL, Yablonka-Reuveni Z, Chamberlain JR and Chamberlain JS. Adeno-associated viral vectors do not efficiently target muscle satellite cells. Mol. Ther. Meth. & Clin. Dev. Sept. 2014, 1;14038. (PMID: 25580445)
- Strakova J, Dean JD, Sharpe K, Meyers TA, Odom GL, and Townsend D. Alpha-dystrobrevin potentiates the function of dystrophin function in the heart by reinforcing dystrophin’s interaction with the membrane. J. Mol. Cell Cardiol. 2014 Nov. 76:106-115. (PMID: 25158611)
- Swiderski K, Shaffer S, Gallis B, Odom GL, Arnett AL, Edgar JS, Baum DM, Chee A, Naim T, Gregorevic, P, Murphy, KT, Goodlett, DR, Lynch GS, and Chamberlain, JS. Phosphorylation within the cysteine-rich region of dystrophin enhances its association with β-dystroglycan and identifies a potential novel therapeutic target for skeletal muscle wasting. Hum Mol Genet. 2014 Dec 20; 23(25):6697-711. (PMID: 25082828)
- Su W, Kang J, Sopher B, Gillespie J, Macarena, AS, Odom GL, Case A, Wang DB, Chamberlain JS, and Garden G. Adeno-associated virus (AAV) vectors mediate efficient gene transduction in cultured neonatal and adult microglia. J. Neurochem. 2016 Jan; 136 Suppl 1:49-62. (PMID: 25708596)
- Odom GL, Kolwicz SC, Nowakowski SG, Moussavi-Harami, F, Flint GV, Gay E, Stuppard R, Gu H, Djukovic D, Chamberlain JS, Raftery D, Tian R, Marcinek DJ, Murry C, and Regnier M. AAV6-mediated Cardiac Specific Over-expression of Ribonucleotide Reductase Enhances Myocardial Contractility. Mol. Ther. 2016 Feb; 24(2):240-50. (PMID: 26388461)
- Whitehead N, Bible K, Kim MJ, Adams M, Odom, GL and Froehner SC. Validation of ultrasonography for non-invasive assessment of diaphragm function in muscular dystrophy. J Physiol. 2016 Dec 15;594(24):7215-27. (PMID: 27570057)
- Bengtsson N, Hall J, Odom G, Phelps MP, Andrus CR, Hawkins DR, Hauschka SD, Chamberlain JC, and Chamberlain JS. Enhanced muscle-specific CRISPR/Cas9 editing of the dystrophin gene ameliorates pathophysiology in a mouse model of DMD. Nature Comm. 2017 Feb; 8:14454.
Reviews
- Odom GL, Gregorevic P, and Chamberlain JS. Viral-mediated gene therapy for the muscular dystrophies: successes, limitations and recent advances. Bioch Biophys Acta. 2007, Feb; 1771(2):243-62. (PMID: 17064882)
- Seto J, Ramos JN, Muir L, Chamberlain JS, and Odom GL. Gene replacement therapies for Duchenne muscular dystrophy using adeno-associated viral vectors. Current Gene Therapy. 2012 Jun 1;12(3):139-51. (PMID: 22533379)
- Bengtsson N, Seto JS, Hall JK, Chamberlain JS, and Odom GL. Progress and Prospects of Gene therapy clinical trials for the muscular dystrophies. Hum Mol Genet. 2016 Apr 15;25(R1):R9-R17. (PMID: 26450518)
- Thompson KS, Odom GL, Murry CE, Mahairas GG, Moussavi-Harami F, Teichman SL, Chan X, Hauschka SD, Chamberlain JS, Regnier M. Overexpression of Ribonucleotide Reductase with Gene Therapy Treats Heart Failure by Activating Cardiac Myosin and Enhancing Myocardial Contractility. JACC 2016 Dec 1; 1(7):666-679.
Research
Deimmunization of human T-cell reactive dystrophin epitopes
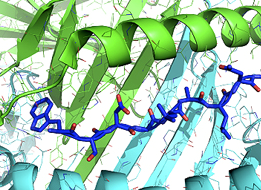 A significant hurdle for DMD gene therapy for many patients is the likelihood of a T-cell mediated immune response against micro-dystrophin (µDys). We seek to avoid this problem via a Rosetta-based protein immuno-silencing & redesign protocol that integrates human immune epitope data, MHC epitope or antigen prediction tools (structure-based design) & proteomic data (sequence-based design) to derive µDys proteins with highly reduced immunogenicity while retaining stability & function. Our proposal builds on preliminary data demonstrating µDys can be redesigned to eliminate immunogenicity of a human T-cell epitope, (previously identified in a clinical trial), while retaining functional properties in dmd mice. Importantly, the algorithm used in our studies predicted T-cell responses to the same epitope, independently of its detection in the clinical trial. The application of T-cell epitope redesign to internally truncated dystrophins also applies to novel junctions in dystrophin resulting from exon-skipping or Crispr/Cas9-based approaches. A significant hurdle for DMD gene therapy for many patients is the likelihood of a T-cell mediated immune response against micro-dystrophin (µDys). We seek to avoid this problem via a Rosetta-based protein immuno-silencing & redesign protocol that integrates human immune epitope data, MHC epitope or antigen prediction tools (structure-based design) & proteomic data (sequence-based design) to derive µDys proteins with highly reduced immunogenicity while retaining stability & function. Our proposal builds on preliminary data demonstrating µDys can be redesigned to eliminate immunogenicity of a human T-cell epitope, (previously identified in a clinical trial), while retaining functional properties in dmd mice. Importantly, the algorithm used in our studies predicted T-cell responses to the same epitope, independently of its detection in the clinical trial. The application of T-cell epitope redesign to internally truncated dystrophins also applies to novel junctions in dystrophin resulting from exon-skipping or Crispr/Cas9-based approaches.
Combinatorial therapies toward alleviating cardiopulmonary deficits in an advanced model of DMD
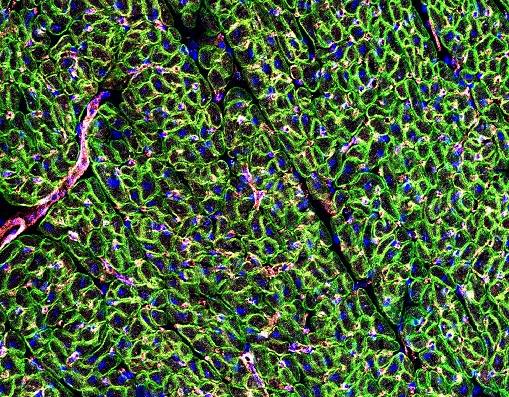 DMD cardiomyopathy is a common feature developing in approximately the second decade of life. As supportive therapies for respiratory function improve, cardiomyopathy, resulting in heart failure, has become a primary cause of death in an ever increasing number of patients. The goal of this project is to provide important information towards a potentially translatable cardiac therapy for DMD patients with advanced disease; likely not responsive toward skeletal muscle gene replacement therapies alone. As such, we aim to comprehensively evaluate and characterize the potential synergy between two promising gene therapy approaches on ameliorating cardiopulmonary performance, independently in young and advanced aged dystrophic dmd mice (a model of end-stage Duchenne dilated cardiomyopathy). Addressing the underlying genetic defect of DMD (the lack of dystrophin), thereby protecting myofibers from further injury, while simultaneously improving the relative magnitude & rate of cardiac contractility via alternative nucleotide therapy (ribonucleotide reductase) with ionotropic enhancement, potentially provides for a unique therapeutic advantage. DMD cardiomyopathy is a common feature developing in approximately the second decade of life. As supportive therapies for respiratory function improve, cardiomyopathy, resulting in heart failure, has become a primary cause of death in an ever increasing number of patients. The goal of this project is to provide important information towards a potentially translatable cardiac therapy for DMD patients with advanced disease; likely not responsive toward skeletal muscle gene replacement therapies alone. As such, we aim to comprehensively evaluate and characterize the potential synergy between two promising gene therapy approaches on ameliorating cardiopulmonary performance, independently in young and advanced aged dystrophic dmd mice (a model of end-stage Duchenne dilated cardiomyopathy). Addressing the underlying genetic defect of DMD (the lack of dystrophin), thereby protecting myofibers from further injury, while simultaneously improving the relative magnitude & rate of cardiac contractility via alternative nucleotide therapy (ribonucleotide reductase) with ionotropic enhancement, potentially provides for a unique therapeutic advantage.
The potential role of dystrophin family protein members within the DGC
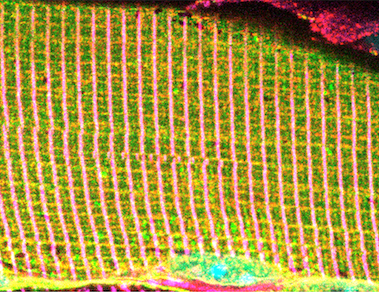 Alpha dystrobrevin (α-Db), a dystrophin superfamily member, is a component of the dystrophin-glycoprotein comlex (DGC) within striated muscles. Decreasing amounts of α-Db from the sarcolemma contributes to severity of disease in several muscular dystrophies including Duchenne muscular dystrophy (DMD). We have shown by whole-body recombinant adeno-associated viral vector (rAAV) gene transfer into α-Db null mice, that a formerly understudied isoform, α-DB3, becomes integrated within the DGC, localizes to costameres, prevents muscle degeneration, & restores diaphragm function. The association of α-Db3 within the DGC appears essential for its function, whose absence likely contributes to sarcolemma fragility & susceptibility to injury in multiple muscular dystrophies. Ongoing efforts are aimed at elucidating novel protein-protein interactions that facilitate retention of α-Db3 to the DGC &/or costameres, which in turn lead to functional benefits and prevention of myopathy in α-Db null mice, even in the presence of numerous key DGC components. Alpha dystrobrevin (α-Db), a dystrophin superfamily member, is a component of the dystrophin-glycoprotein comlex (DGC) within striated muscles. Decreasing amounts of α-Db from the sarcolemma contributes to severity of disease in several muscular dystrophies including Duchenne muscular dystrophy (DMD). We have shown by whole-body recombinant adeno-associated viral vector (rAAV) gene transfer into α-Db null mice, that a formerly understudied isoform, α-DB3, becomes integrated within the DGC, localizes to costameres, prevents muscle degeneration, & restores diaphragm function. The association of α-Db3 within the DGC appears essential for its function, whose absence likely contributes to sarcolemma fragility & susceptibility to injury in multiple muscular dystrophies. Ongoing efforts are aimed at elucidating novel protein-protein interactions that facilitate retention of α-Db3 to the DGC &/or costameres, which in turn lead to functional benefits and prevention of myopathy in α-Db null mice, even in the presence of numerous key DGC components.
 Utrophin (Utrn), an additional dystrophin superfamily member, holds a high degree of sequence similarity to that of dystrophin; so much so that over 70 exons spanning > 900 kb reveals intron/exon structural identity. In normal adult skeletal muscle, Utrn can be found concentrated at the neuromuscular and myotendinous junctions; whereas in newly regenerated myofibers it is also localized to the sarcolemma. Further, in early fetal development Utrn can also be found localized to the sarcolemma in skeletal muscle at ~11 weeks and wanes at ~23 weeks postnatal when expression of dystrophin ensues. This difference in developmental expression resulted in the proposal that Utrn may be the autosomal fetal form of dystrophin. Thus, leading to the hypothesis that Utrn may be able to functionally compensate for dystrophin in DMD patients. Implying a gene therapy approach focusing on the exogenous delivery of µUtrn to DMD muscle fibers could be a viable approach for treating DMD. We have been developing truncated µUtrn vectors capable of being packaged within rAAV that are designed to increase efficacy. Our lab is continuing studies to develop & refine µUtrn function with the goal of providing a long lasting alternative therapeutic for DMD. Utrophin (Utrn), an additional dystrophin superfamily member, holds a high degree of sequence similarity to that of dystrophin; so much so that over 70 exons spanning > 900 kb reveals intron/exon structural identity. In normal adult skeletal muscle, Utrn can be found concentrated at the neuromuscular and myotendinous junctions; whereas in newly regenerated myofibers it is also localized to the sarcolemma. Further, in early fetal development Utrn can also be found localized to the sarcolemma in skeletal muscle at ~11 weeks and wanes at ~23 weeks postnatal when expression of dystrophin ensues. This difference in developmental expression resulted in the proposal that Utrn may be the autosomal fetal form of dystrophin. Thus, leading to the hypothesis that Utrn may be able to functionally compensate for dystrophin in DMD patients. Implying a gene therapy approach focusing on the exogenous delivery of µUtrn to DMD muscle fibers could be a viable approach for treating DMD. We have been developing truncated µUtrn vectors capable of being packaged within rAAV that are designed to increase efficacy. Our lab is continuing studies to develop & refine µUtrn function with the goal of providing a long lasting alternative therapeutic for DMD.
Contact Us
 Mail: Mail:
Odom Laboratory
University of Washington School of Medicine
Department of Neurology
1959 NE Pacific Street
HSB Box# 357720
Seattle, WA 98195
Phone: 206-221-5412
Fax: 206-616-8272
Email: This email address is being protected from spambots. You need JavaScript enabled to view it.
|
