WHAT IS EPIGENETICS?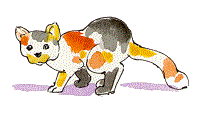
![]() Epigenetics links the fields of genetics and developmental biology.
Epigenetics links the fields of genetics and developmental biology.
![]() Epigenetics is the fundamental biological process by which organisms with two or more different cell types establish patterns of differential gene expression that are stable through cell division. Several interesting examples of epigenetic inheritance are also known for bacteria.
Epigenetics is the fundamental biological process by which organisms with two or more different cell types establish patterns of differential gene expression that are stable through cell division. Several interesting examples of epigenetic inheritance are also known for bacteria.
![]() Mechanisms of epigenetic inheritance include methylation of DNA, modification of histones, binding of transcription factors to chromatin, and the timing of DNA replication. Of these mechanisms, the inheritance of DNA methylation is best understood.
Mechanisms of epigenetic inheritance include methylation of DNA, modification of histones, binding of transcription factors to chromatin, and the timing of DNA replication. Of these mechanisms, the inheritance of DNA methylation is best understood.
![]() Epigenetic processes can be studied in both inherited and somatic human diseases.
Epigenetic processes can be studied in both inherited and somatic human diseases.
![]() The calico cat pictured here provides a good visual example of the epigenetic linkage between genetics and developmental biology. Genetically identical cells in a female cat express either orange or black coat-color genes, depending on whether the paternal or the maternal X-chromosome in that cell is the active X-chromosome. Epigenetic inheritance is stable in this example, as the pattern of X-inactivation is inherited in the clones of cells that give rise to large patches of orange or black fur.
The calico cat pictured here provides a good visual example of the epigenetic linkage between genetics and developmental biology. Genetically identical cells in a female cat express either orange or black coat-color genes, depending on whether the paternal or the maternal X-chromosome in that cell is the active X-chromosome. Epigenetic inheritance is stable in this example, as the pattern of X-inactivation is inherited in the clones of cells that give rise to large patches of orange or black fur.
![]()
EARLY EPIGENETICS RESEARCH FROM THE LAIRD LAB
Charles first presented epigenetic concepts and data analysis at a Gordon Conference in 1986, and published the application of this work to Fragile X syndrome in 1987. In these papers he presented two complementary models explaining the cytogentic and cognitive phenotypes of Fragile X syndrome (Laird et al., 1987; Laird, 1987).
Subsequent research in this area by the Laird Lab and other labs has confirmed several of his early molecular epigenetic predictions concerning CpG-island hypermethylation (Hansen et al., 1992), and late-replication (Hansen et al., 1993) of a critical locus, FMR1. In addition, these complementary models were brought together by his prediction (Laird et al., 1987), later confirmed by Jost and colleagues using chick embryonic cells, that demethylation by a DNA glycosylase requires hemimethylated DNA.
We have extended these findings to broader epigenetic issues such as the extensive mosaicism in DNA methylation among cells and among CpG sites (Stöger et al., 1997), and to the very late DNA replication of many human sequences (Widrow et al., 1998), a finding that challenges the text-book view of the cell cycle.
CURRENT LAB RESEARCH IN EPIGNETICS
![]() Our most recent work on epigenetics is facilitated by our development of three new methods that permit us to determine methylation patterns on both strands of individual DNA molecules (Laird et al., 2004). With our new method of "hairpin-bisulfite PCR", we inferred regional rates of maintenance and de novo methylation for the FMR1 locus in leukocytes that were not subjected to cell culture or other manipulation.
Our most recent work on epigenetics is facilitated by our development of three new methods that permit us to determine methylation patterns on both strands of individual DNA molecules (Laird et al., 2004). With our new method of "hairpin-bisulfite PCR", we inferred regional rates of maintenance and de novo methylation for the FMR1 locus in leukocytes that were not subjected to cell culture or other manipulation.
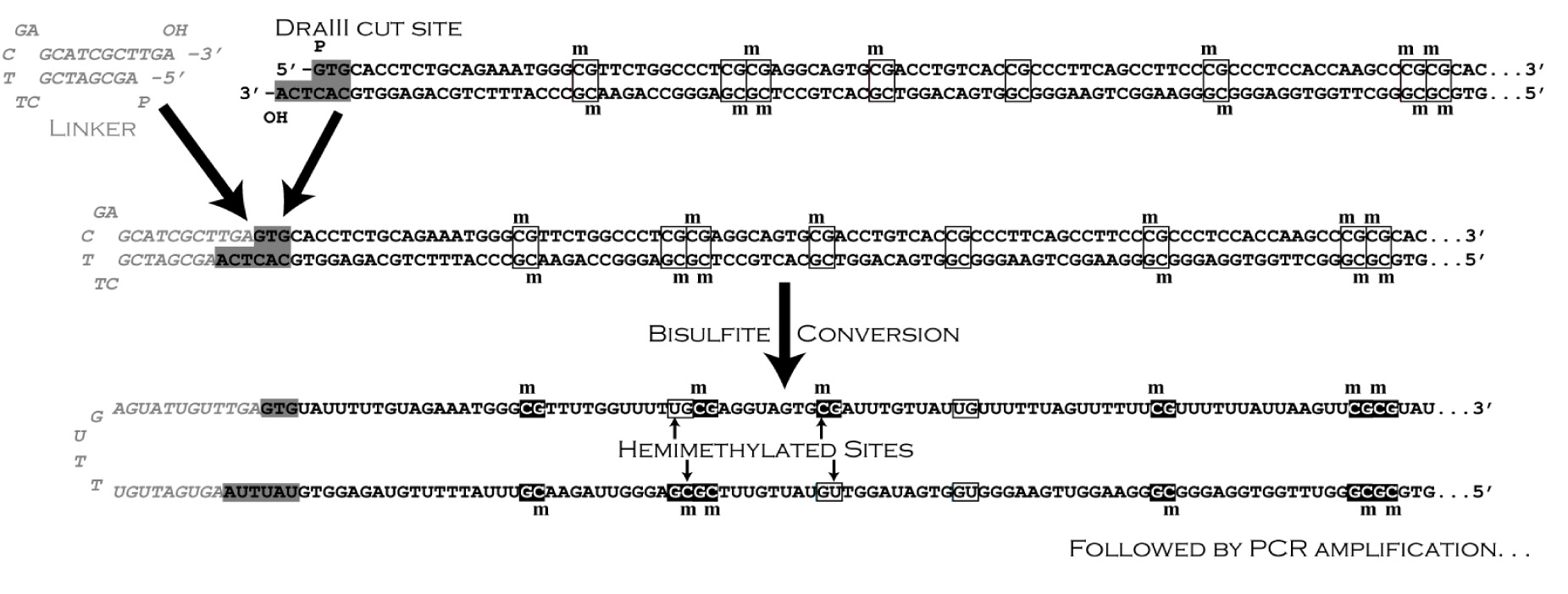
Maintenance and de novo methylation rates were estimated for a portion of the CpG island of FMR1 on the inactive-X chromosome. The active-X allele of FMR1 was strikingly different from the inactive-X allele in having no significant methylation on either strand. Thus, the de novo methylation rate for the inactive X allele must be very close to 0.0 per CpG site per division, with no opportunities for maintenance methylation. We are interested in the question of how two genetically identical alleles in the same female cell can experience either high or near-zero rates of de novo methylation.
![]() We have applied hairpin-bisulfite PCR to human LINE-1 (L1) transposable elements to resolve two controversies in the literature. (Burden et al., 2005). We find that levels of hemimethylation in L1s from a variety of human cell types have low to modest levels of hemimethylation, rather than high levels reported by Woodcock et al. (1997). We also observe that non-CpG cytosine methylation is very rare in two regions of the L1 promoter. Previous methods relied on independent PCR of template and non-template strands, which can lead to amplification and cloning biases. Our new methods are thus useful in analyzing repeated sequences, where the exact complementary strands of individual alleles are held together through bisulfite conversion and PCR amplification.
We have applied hairpin-bisulfite PCR to human LINE-1 (L1) transposable elements to resolve two controversies in the literature. (Burden et al., 2005). We find that levels of hemimethylation in L1s from a variety of human cell types have low to modest levels of hemimethylation, rather than high levels reported by Woodcock et al. (1997). We also observe that non-CpG cytosine methylation is very rare in two regions of the L1 promoter. Previous methods relied on independent PCR of template and non-template strands, which can lead to amplification and cloning biases. Our new methods are thus useful in analyzing repeated sequences, where the exact complementary strands of individual alleles are held together through bisulfite conversion and PCR amplification.
![]() Most recently, a "population-epigenetic" model of DNA methylation has been developed that allows calculation of CpG site-specific methylation rates using our new double-strand methylation data (Genereux et al., 2005). Our initial analysis of several CpG sites of FMR1 suggest that there are significant differences among sites in rates of either maintenance or de novo methylation. With these analytical tools, we can determine whether both of these processes are contributing to these differences.
Most recently, a "population-epigenetic" model of DNA methylation has been developed that allows calculation of CpG site-specific methylation rates using our new double-strand methylation data (Genereux et al., 2005). Our initial analysis of several CpG sites of FMR1 suggest that there are significant differences among sites in rates of either maintenance or de novo methylation. With these analytical tools, we can determine whether both of these processes are contributing to these differences.
![]() A third method that we have recently developed contributes to the authenticity of our methylation data, and allows high confidence in the above conclusions. Molecular barcodes and batch-stamps are now added to the hairpin linker that joins the complementary strands of individual DNA molecules (Miner et al., 2004). This new method allows us to distinguish between valid sequences and those that arise from limited template redundancy or contamination.
A third method that we have recently developed contributes to the authenticity of our methylation data, and allows high confidence in the above conclusions. Molecular barcodes and batch-stamps are now added to the hairpin linker that joins the complementary strands of individual DNA molecules (Miner et al., 2004). This new method allows us to distinguish between valid sequences and those that arise from limited template redundancy or contamination.