The information in this supplement complements our recent analysis of the genome sequence published in the 14 December 2001 issue of Science: Wood, D. W., J. C. Setubal, R. Kaul, D. E. Monks, J. P. Kitajima, V. K. Okura, Y. Zhou, et al. The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science 294 (5550): 2317-2323. The data at this site will be periodically updated to reflect continuing analyses. Please refer any questions to agro@u.washington.edu.
Original supplemental data is available online at the Science website. NOTE: Supplemental data at the Science site utilizes our original gene numbering system which has been subsequently updated. Cross matches between old and new numbering systems are available.
We will host access to supplemental data for the accompanying sequence paper by Goodner et al. (Science 294: (5550): 2323-2328). This data will be available shortly in a version that will be periodically updated by the group at Cereon Genomics. This data is also available at Science Online. This data was provided by Dr. Steven Slater of Cereon Genomics, please address any questions to steven.c.slater@cereon.com.
Genbank accession numbers
Sequencing
Southern analysis of linear chromosome
Origin of replication
Annotation
Paralogous families
GC content
Codon usage
Insertion sequences analysis
Phylogenetic analyses
COG
Phylogenetic trees
16S DNA
Protein
(manuscript fig. 2B/C)
Whole genome comparisons and unique genes
nucleotide alignments
protein comparisons
Top
blast hit analysis (manuscript fig. 2A)
Bi-directional
Best Hit (BBH) analysis (manuscript fig. 3)
Ortholog
analysis (manuscript fig. 1, Table 2)
Unique
genes
Transporter analysis
Regulatory families
Metabolic pathway analysis
References
![]()
The sequence has been deposited at Genbank under the following accession
numbers: Circular chromosome (AE008688), linear chromosome (AE008689),
pAtC58 (AE008687), pTiC58 (AE008690). These sequences will be available
publicly following the publication of the manuscript.
Agrobacterium tumefaciens strain C58 was sequenced using
standard DNA sequencing protocols and data collection tools, and was based
on the shotgun approach (1). Two small-insert libraries were constructed
in pUC18 with an average insert length of 2.0 kb and 5.0 kb respectively.
DNA was physically sheared by nebulization, and size fractionated by agarose
gel electrophoresis. DNA was extracted from 96-well plate cultures
using the Qiagen (Chatsworth, CA) R.E.A.L. method. Cosmid and fosmid libraries,
with inserts ranging between 40 and 50-kb, were also generated. All clones
were sequenced from both ends using M13 universal primers. Sequencing reactions
were performed with dRhodamine Dye terminator or Big-dye terminator chemistry
(Applied Biosystems Prism Dye Terminator Reaction Ready) and run on ABI
377-XL DNA sequencers or ABI 3700 CE sequencers.
A total of 135,703 reads (132,226 small insert shotgun reads, 1,359-cosmid and 2,118-fosmid end-sequencing reads) were generated. Sequences were assembled and viewed using the phred/phrap/consed software (http://www.phrap.org). The assembly was carried out on a 667-MHZ dual processor Compaq alpha computer with 8 GB of memory. The initial sequence assembly took about nine hours. To facilitate opening and viewing the genome assemblies in consed, a phd.ball file from all phd files was created. The creation of the phd.ball file reduced the time to open the genome assembly in consed to less than 15 min. The initial assembly provided 8.68X Q20 sequence coverage (Q20: phred error rate < 1%) (2), with 99.4% genome coverage. The genome was finished using the autofinish tool of consed (16). Periodically the sequences were reassembled with each assembly requiring about five-and-a half-hours of processing time. The final assembly contained 131,299 reads including 3,730 reads from autofinish and advanced finishing experiments. Five gross misassemblies were identified by analysis of the paired ends of fosmid end-sequences. They were all due to the presence of nearly identical repeat sequences in the genome. These repeat sequences included four copies of the 6.7-Kb ribosomal DNA (two copies each in circular and linear replicons) and three copies of a 1,598 base sequence (one copy in each of the circular, linear and pATC58 replicons). Unique fosmid clones that spanned misassembled regions were selected and sequenced to 8X Q20 coverage. The final validation of the sequence assembly was achieved by comparing the restriction fragment digest of 269 fosmid clones (providing nearly continuous 2X-genome coverage) with the computational restriction digests of the final assembly. An independent validation of the sequence quality was provided by comparison of the final C58 sequence based on the whole genome assembly with the independently finished sequence of two randomly chosen fosmid clones. The two quality-control fosmid clones spanned a combined length of 81,801 bases, accounting for 1.44% of the genome. There were no discrepancies between the whole genome assembly and the sequence of the two quality-control fosmid clones.
Genomic DNA was prepared from A. tumefaciens strain C58,
including protease treatment, as previously described (3). Restriction
sites were chosen and digests performed such that small fragments of defined
size were generated from each end. Three restriction sites were chosen
at each end, for a total of six digests. Southern blot analysis was performed
on the digested DNA using radiolabelled probes specific for each end. Resultant
band sizes, consistent with that predicted from the linear chromosome sequence
data, indicated that complete sequence had been obtained to within a 40-bp
agarose gel resolution.
The putative origin of replication for the circular chromosome was
determined based on gene cluster conservation (4) and confirmed by GC-skew
[(G-C)/(G+C)] analysis (10 kb sliding window). Base 1 of this sequence
was chosen to be an arbitrary base midway between genes AtC58-1970 (ortholog
to RP001) and AtC58-1971 (hemE). The genes around this putative
origin are the following:
-parB -parA -gidB -gidA -thdF +hypo(*) +hypo(*) -TFrho -RP883 -hemH(*) -hemE ORIGIN +RP001 +maf +aroE +hypo/coaE +dnaQ
The signs +/- denote strand. The order and content depicted are those found in Caulobacter crescentus, as described in (4). Those marked with (*) are not found in Agrobacterium tumefaciens C58 at this location. Sinorhizobium meliloti has exactly the same genes as A. tumefaciens in the region depicted, and its origin has been chosen at the point indicated above.
The origin of replication of the AT plasmid is thought to be just upstream of the repABC operon (5). We chose base 1 of this sequence as an arbitray base midway between genes AtC58-436 (repA) and AtC58-435 (hypothetical). The origin of replication of the Ti plasmid is also thought to be near its repC gene, but we chose base 1 as the first base of the left T-border sequence, consistent with previously published Ti plasmid sequences from other A. tumefaciens strains.
The origin of replication of the linear chromosome is thought to be close to its center (which is near position 1,037,780). There is a repABC operon close to the center (repA start is at 1,026,567), and at position 1,076,640 there is a major signal inversion of the GC-skew curve (10 kb sliding window).
Annotation was carried out on an annotation system and interface
developed at the Bioinformatics
Laboratory of the University of Campinas, Brazil.
Putative genes were identified using BLASTX (6) and Glimmer (7). Protein function was predicted based on comparisons to sequences in the public databases Genbank (using BLAST (6)), PFAM (8), and COG (9). These comparisons were used as auxiliary tools by human curators, who annotated each gene. The curators assigned predicted proteins to one of 18 functional categories modified from those described by Riley (10). RNA species were identified using BLASTN (6) and tRNAscan-SE (11). We thank K. Williams for assistance in identifying the tmRNA borders.
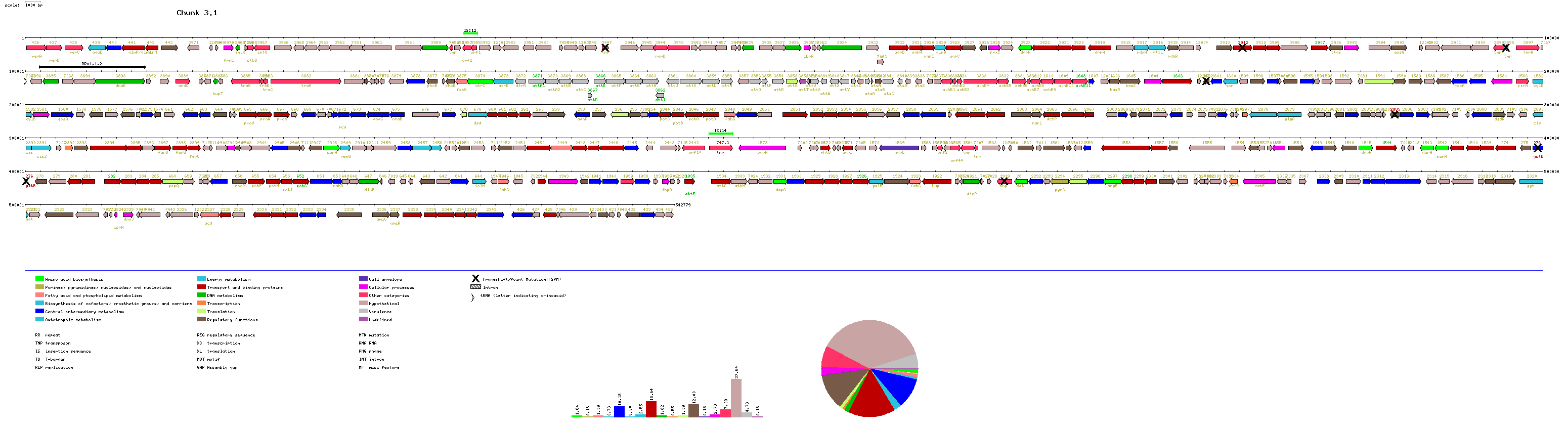
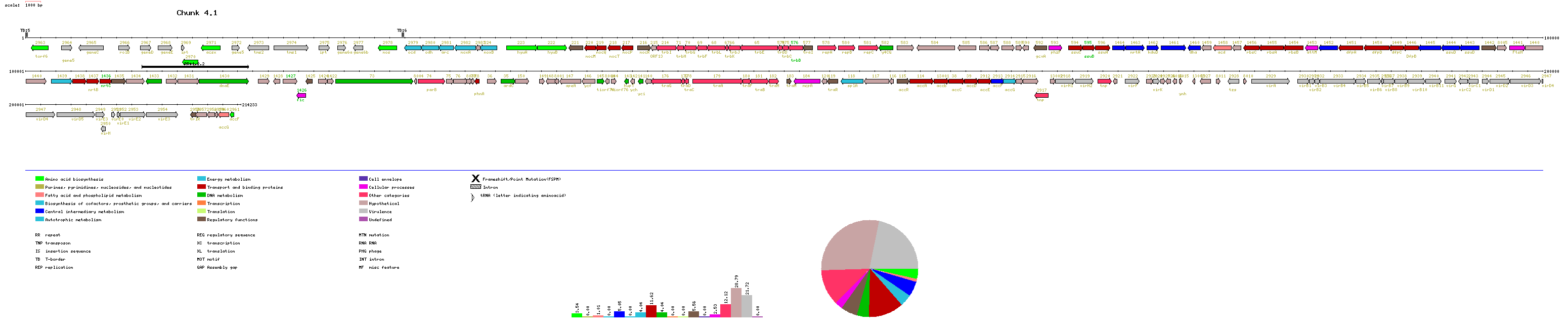
A text file containing the complete list of A. tumefaciens protein-coding genes separated in categories is available. This file is 586 kilobytes long. Gene maps for each replicon are available from the following links:
A gene from A. tumefaciens (the query) was said to be paralogous
to another A. tumefaciens gene (the subject) if a BLASTP (6) of
the query against the subject resulted in a hit with e-value less than
or equal to e-5 and query and subject coverage were both at least 60%,
with some additional clustering processing. The BLAST subject database
in this case was the entire A. tumefaciens proteome. A complete
list of the paralogous
families and their membership is available.
Manuscript figure 1 contains information of GC content on a gene-by-gene
basis. Here we present GC content graphs using a 5 kb sliding window. Datapoints
on each graph reflect the GC content of the window centered at that point.
Codon frequencies were computed using the codonw program written
by John Peden: http://molbiol.ox.ac.uk/cu.
A
comparison
of codon usage frequencies for the entire genome, each of the replicons
and the T-DNA, vir genes and AT island is available. (Note: Please
use the magnify function of Acrobat reader to view the file details since
the file size is quite large and it opens in a small format window).
This table is divided into 8 sections: the whole genome, one section for each of the replicons, the AT island, T-DNA, and the genes of the vir region. For each section, the table lists the frequencies at which codons occur within groups of the same aminoacid. For example, if an aminoacid has 6 codons, the relative frequencies for its codons add up to 6. This means that codons with relative frequencies greater than 1 are overrepresented with respect to uniform usage. Similarly, codons with relative frequencies less than 1 are underrepresented. Therefore, relative frequencies reflect biases in codon usage. Two simple comparisons were made in an attempt to compare codon usage between subsets of genes in the genome with the codon usage in the genome as a whole. First, codon usage frequencies for individual codons were expressed as a percentage of the frequency in the genome as a whole. Second, the absolute difference between these same two frequencies was determined.
Graphs showing codon bias on all 4 replicons are also available. They were done following the methodology proposed by Karlin (21) and using a sliding window of 20 kb. In this methodology codon bias is found by comparing the codon usage in the genes in each window to the codon usage of the genome as a whole.
Insertion Sequences (IS) were identified manually using tools available
at the Insertion Sequence database (http://www-IS.biotoul.fr/is.html).
COG analysis
Predicted proteins of A. tumefaciens and S. meliloti were run through the COGnitor program (9). The results were combined with the COG database available from NCBI, which contained 44 complete genomes at the time of analysis (August, 2001). A comparison of the major COG groups for all sequenced organisms sorted by genome size is available.
Phylogenetic trees
16S rDNA trees
For analysis of rDNA sequences, an alignment of small subunit rDNA sequences for alpha-proteobacteria was downloaded from the European Large Subunit Ribosomal RNA Database (14). The 16S rDNA sequences for the three Rhizobia (Sinorhizobium meliloti, Mesorhizobium loti, and Caulobacter crescentus) for which complete genomes are available were added to this alignment manually. Phylogenetic trees were generated from this alignment (after redundant and incomplete sequences were removed and poorly aligned columns were excluded) and from selected subsets of the alignment using parsimony, distance, and likelihood methods available in the PAUP (http://paup.csit.fsu.edu/about.html) program. The 16S rDNA tree containing A.tumefaciens, S. meliloti, M. loti and related species is available.
Protein sequence trees
For analysis of protein sequences, a set of proteins was chosen (RecA, EF-Tu, Ef-G, HSP70, HSP60, RpoA, RpoB, RpoC, some ribosomal proteins, DnaJ) for which molecular systematics has been shown to be reasonably reliable and for which homologs are available in all complete genomes of free living bacteria. For each protein, a multiple sequence alignment was generated including all homologs. A tree was generated from these alignments, after ambiguously aligned positions were excluded, using PAUP distance methods and a distance calculation based on PAM matrices. Trees in the manuscript figure 2B and 2C were made using these same methods. Trees for each of the other proteins listed are available by request from Jonathan Eisen at The Institute for Genomic Research.
Nucleotide alignments
The A. tumefaciens genome was compared at the nucleotide level to other genomes using MUMmer (15) with default parameter values. The following nucleotide comparisons are available:
A. tumefaciens circular chromosome X S. meliloti chromosome A. tumefaciens circular chromosome X M. loti chromosome S. meliloti chromosome X M. loti chromosome
Note:The M. loti chromosome sequence was circularly shifted in these comparisons. Base 1 here corresponds to base 3572980 in the original sequence.
Top BLAST hit analysis
Manuscript figure 2A was generated by comparing predicted proteins of A. tumefaciens with proteins from all published complete genomes using fasta3 (13). Top blast hits for all genes were cataloged and the percent of top BLAST hits from each organism was calculated. Top hits for each replicon are also available.Bi-directional Best Hit (BBH) analysis
Bi-directional best hits were determined using the following approach. A bi-directional best hit (BBH) is a pair (p1,p2) of proteins, p1 in genome A, and p2 in genome B, such that when the proteome of A is BLASTed against the proteome of B, p2 comes out as the best hit for p1, and when B is BLASTed against A p1 comes out as the best hit for p2. The cutoff e-value used was 10-4.Ortholog analysisBBHs were used to generate the proteome alignments in manuscript figure 3 and those shown below. The graphs in the following list show whole proteome comparisons of the A. tumefaciens linear replicon with the chromosomes of S. meliloti and M. loti. As in manuscript figure 3, each data point is a BBH. In these graphs, the regions of gene order conservation can be seen as strings of consecutive or nearly consecutive points. The graphs are enlarged to show two extensive regions of gene order conservation. The first graph shows the conservation with respect to the S. meliloti chromosome. In S. meliloti, these genes are located in a section that is not colinear with the A. tumefaciens circular chromosome. The second graph shows that roughly the same two regions are conserved with respect to the M. loti chromosome.
A detailed view of one of the regions with extensive gene order conservation mentioned above can be seen here. This table was generated by a program described in (12).The numbers of orthologs shown in manuscript table 2 were determined using the following methods. These same data were used to color code the individual open reading frames in manuscript figure 1. This definition of orthologs was used instead of BBHs to allow the orthologs of paralogs to be found. Two proteins were considered orthologs if their BLASTP alignment covered at least 60% of each protein at an expect value of less than or equal to 10-5. Proteins that did not match these criteria were considered non-orthologous. Manuscript figure 1 was generated by a program adapted from the genome_plot program. Genome_plot was written and kindly sent to us by Rene Gibson, from Genome Therapeutics Corporation.Unique genesUnique genes were defined in the following manner. A protein p from genome A was considered unique with respect to genome B if a BLASTP using p as query against the proteome of B yielded no hits at a threshold expect value of 10-3. Here is a list of unique genes in A. tumefaciens with respect to S. meliloti and M. loti.
Transporters were identified using BLASTP (6) and HMM-based searches
against a database of known and putative membrane transport proteins and
classified into families based on the TC system as previously described
(17). Here is a complete
description of A. tumefaciens transporter predictions. See http://www-biology.ucsd.edu/~ipaulsen/transport
for additional details on the methods.
Methods similar to those employed in the Pseudomonas aeruginosa
genome project (18) were used to define the regulatory motifs present in
the A. tumefaciens, S. meliloti, M. loti, C. crescentus,
and
P.
aeruginosa genomes. Regulatory family models were extracted from the
PFAM 6.6 database (8), and used to generate a local database. This database
was then searched using HMMER 2.2g (HMMER User's guide: http://hmmer.wustl.edu)
using as queries the predicted proteins in the genomes of each of these
organisms. A motif was assigned if the search resulted in a match
with an expect value less than or equal to 10-4. Sensor/response
regulator hybrids were defined as proteins which contained both response
regulator and sensor kinase motifs. Regulatory proteins may have more than
one motif. The numbers of regulatory
motifs found in the A. tumefaciens, S. meliloti, M. loti, C. crescentus
and P. aeruginosa genomes are shown. The distribution
among each of the A. tumefaciens replicons is also available.
We analyzed the metabolic pathways of Agro with the PathoLogic program
(19) to assess the evidence for the presence in A. tumefaciens of
pathways in the MetaCyc pathway database (20). The analysis detected
178 metabolic pathways, containing 755 reaction steps, of which 467 steps
had enzymes assigned, and 288 lack enzyme assignments. To assess
the presence or absence of a pathway, the analysis emphasized the presence
of enzymes that are unique to a pathway, to decrease the likelihood of
being misled by the many enzymes that are shared among multiple pathways.
The complete results of this analysis are available at: http://ecocyc.org:1555/AGRO/organism-summary?object=AGRO. The data for metabolic pathways was assembled by our collaborators at SRI International, Drs. Peter Karp and Pedro Romero.
1. R. D. Fleischmann et al., Science 269, 496-512. (1995).
2. B. Ewing, L. Hillier, M. C. Wendl, P. Green, Genome Res 8, 175-85.
(1998).
3. T. Maniatis, E. F. Fritsch, J. Sambrook, Molecular cloning: A laboratory
manual (Cold Spring Harbor Laboratory Press, Plainview, NY, 1989).
4. A. K. Brassinga, R. Siam, G. T. Marczynski, J Bacteriol 183, 1824-9.
(2001).
5. M. A. Ramirez-Romero, N. Soberon, A. Perez-Oseguera, J. Tellez-Sosa,
M. A. Cevallos, J Bacteriol 182, 3117-24. (2000).
6. S. F. Altschul et al., Nucleic Acids Res 25, 3389-402. (1997).
7. A. L. Delcher, D. Harmon, S. Kasif, O. White, S. L. Salzberg, Nucleic
Acids Res 27, 4636-41. (1999).
8. A. Bateman et al., Nucleic Acids Res 28, 263-6. (2000).
9. R. L. Tatusov et al., Nucleic Acids Res 29, 22-28. (2001).
10. M. Riley, Microbiol Rev 57, 862-952. (1993).
11. T. M. Lowe, S. R. Eddy, Nucleic Acids Res 25, 955-64. (1997).
12. J.C. Setubal, N. F. Almeida Jr., DIMACS Workshop on whole genome
comparison, Rutgers University. (2001).
13. W. R. Pearson, Methods Mol Biol 132, 185-219. (2000).
14. J. Wuyts, P. De Rijk, Y. Van de Peer, T. Winkelmans, R. De Wachter,
Nucleic Acids Res 29, 175-7. (2001).
15. A. L. Delcher et al., Nucleic Acids Res 27, 2369-76. (1999).
16. D. Gordon, C. Desmarais, P. Green, Genome Res 11, 614-25. (2001).
17. I. T. Paulsen, L. Nguyen, M. K. Sliwinski, R. Rabus, M. H. Saier,
Jr., J Mol Biol 301, 75-100. (2000).
18. C. K. Stover et al., Nature 406, 959-64. (2000).
19. P. D. Karp, M. Krummenacker, S. Paley, J. Wagg, Trends Biotechnol
17, 275-81. (1999).
20. P. D. Karp et al., Nucleic Acids Res 28, 56-9. (2000).
21. S. Karlin, Trends Microbiol 9, 335-343. (2001)
This website and all its contents are the property of the University of Washington. Copyright 2001.
please refer any questions or comments to agro@u.washington.edu
Web design and maintenance: Derek Wood
{kind=link}
{kind=link}
{kind=link}
{kind=link}