Paper of the Month | 2005
October | top
Cutoff size need not strongly influence molecular dynamics results on solvated polypeptides
Beck D.A.C., Armen R.S., and Daggett V.
Biochemistry 45: 609-616, 2005
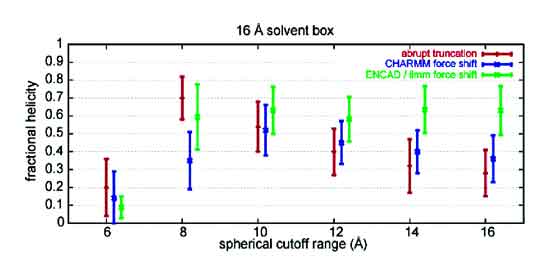
The correct treatment of van der Waals and electrostatic nonbonded interactions in molecular force fields is essential for performing realistic molecular dynamics (MD) simulations of solvated polypeptides. The most computationally tractable treatment of nonbonded interactions in MD utilizes a spherical distance cutoff (typically, 8-12 Â) to reduce the number of pairwise interactions. In this work, we assess three spherical atom-based cutoff approaches for use with all-atom explicit solvent MD: abrupt truncation, a CHARMM-style electrostatic shift truncation, and our own force-shifted truncation. The chosen system for this study is an end-capped 17-residue alanine-based α-helical peptide, selected because of its use in previous computational and experimental studies. We compare the time-averaged helical content calculated from these MD trajectories with experiment. We also examine the effect of varying the cutoff treatment and distance on energy conservation. We find that the abrupt truncation approach is pathological in its inability to conserve energy. The CHARMM-style shift truncation performs quite well but suffers from energetic instability. On the other hand, the force-shifted spherical cutoff method conserves energy, correctly predicts the experimental helical content, and shows convergence in simulation statistics as the cutoff is increased. This work demonstrates that by using proper and rigorous techniques, it is possible to correctly model polypeptide dynamics in solution with a spherical cutoff. The inherent computational advantage of spherical cutoffs over Ewald summation (and related) techniques is essential in accessing longer MD time scales.
November | top
Ensemble versus single-molecule protein unfolding
Day R. and Daggett V.
Proceedings of the National Academy of Sciences USA 102: 13445-13450, 2005
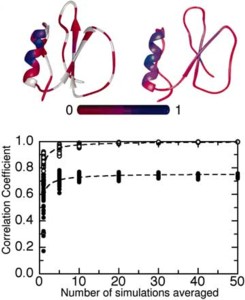
Molecular dynamics (MD) simulations are the classic single-molecule "experiments," providing atomic-resolution structural and dynamic information. However, the single-molecule nature of the technique has also been its shortcoming, with frequent criticisms of sampling inadequacies and questions regarding the ensemble behavior of large numbers of molecules. Given the increase in computer power, we now address this issue by performing a large number of simulations and comparing individual and ensemble properties. One hundred independent MD simulations of the protein chymotrypsin inhibitor 2 were carried out for 20 ns each at 498 K in water to more fully describe the potentially diverse routes of protein unfolding and investigate how representative a single trajectory can be. Rapid unfolding was observed in all cases with the trajectories distributed about an average "ensemble" path in which secondary and tertiary structure was lost concomitantly, with tertiary structure loss occurring slightly faster. Individual trajectories did, however, sample conformations far from the average path with very heterogeneous time-dependent properties. Nevertheless, all of the simulations but one followed the average ensemble pathway, such that a small number of simulations (5-10) are sufficient to capture the average properties of these states and the unfolding pathway.
December | top
Characterization of a possible amyloidogenic precursor in glutamine-repeat neurodegenerative disease
Armen R.S., Bernard B.M., Day R., Alonso D.O.V., and Daggett V.
Proceedings of the National Academy of Sciences USA 102: 13433-13438, 2005
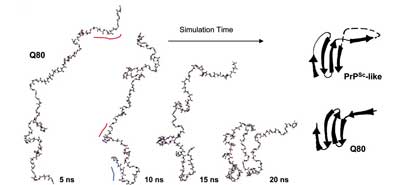
Several neurodegenerative diseases are linked to expanded repeats of glutamine residues, which lead to the formation of amyloid fibrils and neuronal death. The length of the repeats correlates with the onset of Huntington's disease, such that healthy individuals have <38 residues and individuals with >38 repeats exhibit symptoms. Because it is difficult to obtain atomic-resolution structural information for poly(L-glutamine) (polyQ) in aqueous solution experimentally, we performed molecular dynamics simulations to investigate the conformational behavior of this homopolymer. In simulations of 20-, 40-, and 80-mer polyQ, we observed the formation of the "α-extended chain" conformation, which is characterized by alternating residues in the αL and αR conformations to yield a sheet. The structural transition from disordered random-coil conformations to the α-extended chain conformation exhibits modest length and temperature dependence, in agreement with the experimental observation that aggregation depends on length and temperature. We propose that fibril formation in polyQ may occur through an α-sheet structure, which was proposed by Pauling and Corey [Pauling, L. & Corey, R. B. (1951) Proc. Natl. Acad. Sci. USA 37, 251-256]. Also, we propose an atomic-resolution model of how the inhibitory peptide QBP1 (polyQ-binding peptide 1) may bind to polyQ in an α-extended chain conformation to inhibit fibril formation.