Paper of the Month | 2006
January | February | March | April | May | June | July | September
January | top
Local environmental effects on the structure of the prion protein
DeMarco M.L. and Daggett V.
Comptes Rendus Biologies 328: 847-862, 2005
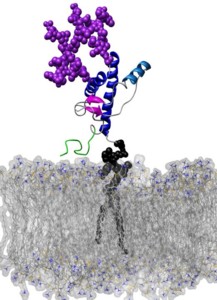
Prion diseases are neurodegenerative diseases causally linked to the partial unfolding and subsequent misfolding and aggregation of the prion protein (PrP). While most proteins fold into a single low energy state, PrP can fold into two distinct isoforms. In its innocuous state, denoted as PrPC, the protein has predominantly α-helical secondary structure, however, PrPC can misfold into an isoform rich in extended structure capable of forming toxic and infectious aggregates. While prion disease is believed to be a protein-only disease, one not requiring any non-protein elements for propagation, the different environments the protein finds itself in vivo likely influence its ability to misfold and aggregate. In this review we will examine various molecules, covalent modifications and environments PrP faces in vivo and the effect they have on PrP's local environment and, potentially, conformation. Included in this discussion are: (1) pH, (2) carbohydrates, (3) lipid membranes, (4) metal ions, and (5) small molecules.
February | top
Characterization of Two Distinct β2-Microglobulin Unfolding Intermediates that May Lead to Amyloid Fibrils of Different Morphology
Armen R.S. and Daggett V.
Biochemistry 44: 16098-16107, 2005
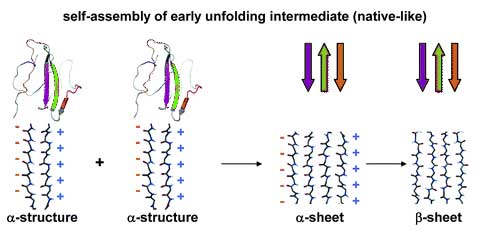
The self-assembly of β2-microglobulin into fibrils leads to dialysis-related amyloidosis. pH-mediated partial unfolding is required for the formation of the amyloidogenic intermediate that then self-assembles into amyloid fibrils. Two partially folded intermediates of β2-microglobulin have been identified experimentally and linked to the formation of fibrils of distinct morphology, yet it remains difficult to characterize these partially unfolded states at high resolution using experimental approaches. Consequently, we have performed molecular dynamics simulations at neutral and low pH to determine the structures of these partially unfolded amyloidogenic intermediates. In the low-pH simulations, we observed the formation of α-sheet structure, which was first proposed by Pauling and Corey. Multiple simulations were performed, and two distinct intermediate state ensembles were identified that may account for the different fibril morphologies. The predominant early unfolding intermediate was native-like in structure, in agreement with previous NMR studies. The late unfolding intermediate was significantly disordered, but it maintained an extended elongated structure, with hydrophobic clusters and residual α-extended chain strands in specific regions of the sequence that map to amyloidogenic peptides. We propose that the formation of α-sheet facilitates self-assembly into partially unfolded prefibrillar amyloidogenic intermediates.
March | top
The 108M Polymorph of Human Catechol O-Methyltransferase Is Prone to Deformation at Physiological Temperatures
Rutherford K., Bennion B.J., Parson W.W., and Daggett V.
Biochemistry 45: 2178-2188, 2006
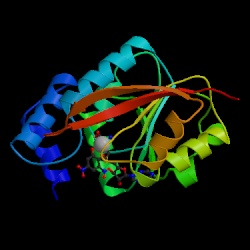
The human gene for catechol O-methyltransferase (COMT) contains a common polymorphism that results in substitution of methionine (M) for valine (V) at residue 108 of the soluble form of the protein. While the two proteins have similar kinetic properties, 108M COMT loses activity more rapidly than 108V COMT at 37°C. The cosubstrate S-adenosylmethionine (SAM) stabilizes the activity of 108M COMT at 40°C. The 108M allele has been associated with increased risk for breast cancer, obsessive-compulsive disorder, and aggressive and highly antisocial manifestations of schizophrenia. In the current work, we have constructed homology models for both human COMT polymorphs and performed molecular dynamics simulations of these models at 25, 37, and 50°C to explore the structural consequences of the 108V/M polymorphism. The simulations indicated that replacing valine with the larger methionine residue led to greater solvent exposure of residue 108 and heightened packing interactions between M108 and helices α2, α4 (especially with R78), and α5. These altered packing interactions propagated subtle changes between the polymorphic site and the active site 16 Å away, leading to a loosening of the active site. At physiological temperature, 108M COMT sampled a larger distribution of conformations than 108V. 108M COMT was more prone to active-site distortion and had greater overall, and SAM binding site, solvent accessibility than 108V COMT at 37°C. Similar structural perturbations were observed in the 108V protein only at 50°C. Addition of SAM tightened up the cosubstrate pocket in both proteins and prevented the altered packing at the polymorphic site in 108M COMT.
April | top
Importance of Context in Protein Folding:
Secondary Structural Propensities versus Tertiary Contact-Assisted Secondary Structure Formation
Scott K.A., Alonso D.O.V., Ping Y., and Daggett V.
Biochemistry 45: 24153-4163, 2006
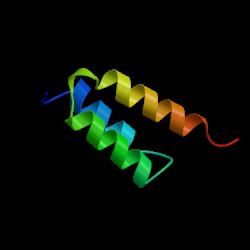
Molecular dynamics simulations can be used to reveal the detailed conformational behaviors of peptides and proteins. By comparing fragment and full-length protein simulations, we can investigate the role of each peptide segment in the folding process. Here, we take advantage of information regarding the helix formation process from our previous simulations of barnase and protein A as well as new simulations of four helical fragments from these proteins at three different temperatures, starting with both helical and extended structures. Segments with high helical propensity began the folding process by tethering the chain through side chain interactions involving either polar interactions, such as salt bridges, or hydrophobic staples. These tethers were frequently nonnative (i.e., not i → i + 4 spacing) and provided a scaffold for other residues, thereby limiting the conformational search. The helical structure then propagated on both sides of the tether. Segments with low stability and propensity formed later in the folding process and utilized contacts with other portions of the protein when folding. These helices formed via a tertiary contact-assisted mechanism, primarily via hydrophobic contacts between residues distant in sequence. Thus, segments with different helical propensities appear to play different roles during protein folding. Furthermore, the active role of nonlocal side chains in helix formation highlights why we must move beyond simple hierarchical models of protein folding.
May | top
The Folding Pathway of Spectrin R17 from Experiment and Simulation:
Using Experimentally Validated MD Simulations to Characterize States Hinted at by Experiment
Scott K.A., Randles L.G., Moran S.J., Daggett V., and Clark J.
Journal of Molecular Biology 359: 159-173, 2006
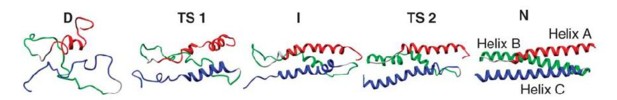
We present an experimental and computational analysis of the folding pathway of the 17th domain of chicken brain α-spectrin, R17. Wild-type R17 folds in a two-state manner and the chevron plot (plot of the logarithm of the observed rate constant against concentration of urea) shows essentially linear folding and unfolding arms. A number of mutant proteins, however, show a change in slope of the unfolding arm at high concentration of denaturant, hinting at complexity in the folding landscape. Through a combination of mutational studies and high temperature molecular dynamics simulations we show that the folding of R17 can be described by a model with two sequential transition states separated by an intermediate species. The rate limiting transition state for folding in water has been characterized both through experimental Φ-value analysis and by simulation. In contrast, a detailed analysis of the transition state predicted to dominate under highly denaturing conditions is only possible by simulation.

July | top
Φ-Analysis at the Experimental Limits: Mechanism of β-Hairpin Formation
Petrovich M., Jonsson A.L., Ferguson N., Daggett V., and Fersht A.R.
Journal of Molecular Biology 360: 865-881, 2006
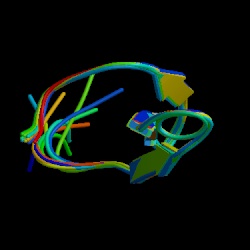
The 37-residue Formin-binding protein, FBP28, is a canonical three-stranded β-sheet WW domain. Because of its small size, it is so insensitive to chemical denaturation that it is barely possible to determine accurately a denaturation curve, as the transition spans 0-7 M guanidinium hydrochloride (GdmCl). It is also only marginally stable, with a free energy of denaturation of just 2.3 kcal/mol at 10°C so only small changes in energy upon mutation can be tolerated. But these properties and relaxation times for folding of 25 μs-400 μs conspire to allow the rapid acquisition of accurate and reproducible kinetic data for Φ-analysis using classical temperature-jump methods. The transition state for folding is highly polarized with some regions having Φ-values of 0 and others 1, as readily seen in chevron plots, with Φ-values of 0 having the refolding arms overlaying and those of 1 the unfolding arms superimposable. Good agreement is seen with transition state structures identified from independent molecular dynamics (MD) simulations at 60, 75, and 100°C, which allows us to explore further the details of the folding and unfolding pathway of FBP28. The first β-turn is near native-like in the transition state for folding (experimental) and unfolding (MD and experiment). The simulations show that there are transient contacts between the aromatic side-chains of the β-strands in the denatured state and that these interactions provide the driving force for folding of the first β-hairpin of this three-stranded sheet. Only after the backbone hydrogen bonds are formed between β1 and β2 does a hydrogen bond form to stabilize the intervening turn, or the first β-turn.
September | top
α-Sheet: The toxic conformer in amyloid diseases?
V. Daggett
Accounts of Chemical Research 39: 594-602, 2006
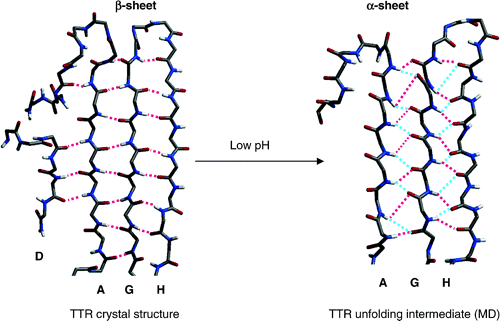
A novel secondary structure, the α-sheet, was identified through molecular dynamics (MD) simulations of various proteins associated with amyloid diseases under amyloidogenic conditions. The structure was first predicted by Pauling and Corey, and it has been directly observed in crystal structures of "nonnatural peptides." There are occurrences of α-strands and α-sheets in the Protein Data Bank, but they are rare. We propose that α-sheet is formed during the conformational changes associated with amyloidosis and that it may represent the toxic conformer. Here, structural properties of the α-sheet, background information, and experimental support for this novel structure are presented. Finally we speculate about the possible role of this conformation in disease.