Paper of the Month | 2007
January | February | March | April | May | October | November | December
January | top
Structural Properties of Prion Protein Protofibrils and Fibrils:
An Experimental Assessment of Atomic Models
DeMarco M.L., Silveira J., Caughey B., and Daggett V.
Biochemistry 45: 15573-15582, 2007
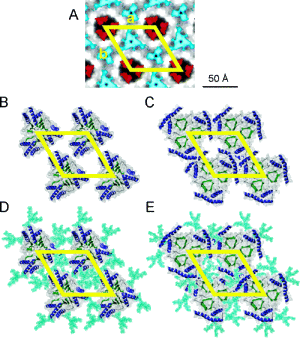
Decades after the prion protein was implicated in transmissible spongiform encephalopathies, the structure of its toxic isoform and its mechanism of toxicity remain unknown. By gathering available experimental data, albeit low resolution, a few pieces of the prion puzzle can be put in place. Currently, there are two fundamentally different models of a prion protofibril. One has its building blocks derived from a molecular dynamics simulation of the prion protein under amyloidogenic conditions, termed the spiral model. The other model was constructed by threading a portion of the prion sequence through a β-helical structure from the Protein Data Bank. Here we compare and contrast these models with respect to all of the available experimental information, including electron micrographs, symmetries, secondary structure, oligomerization interfaces, enzymatic digestion, epitope exposure, and disaggregation profiles. Much of this information was not available when the two models were introduced. Overall, we find that the spiral model is consistent with all of the experimental results. In contrast, it is difficult to reconcile several of the experimental observables with the β-helix model. While the experimental constraints are of low resolution, in bringing together the previously disconnected experiments, we have developed a clearer picture of prion aggregates. Both the improved characterization of prion aggregates and the existing atomic models can be used to devise further experiments to better elucidate the misfolding pathway and the structure of prion protofibrils.
February | top
Folding mechanisms of proteins with high sequence identity but different folds
Scott K.A. and Daggett V.
Biochemistry 46: 1545-1556, 2007
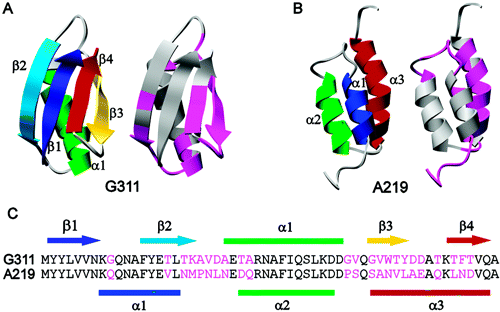
The problem of how a protein folds from a linear chain of amino acids to the three-dimensional structure necessary for function is often investigated using proteins with a low degree of sequence identity that adopt different folds. The design of pairs of proteins with a high degree of sequence identity but different folds offers the opportunity for a complementary study; in two highly similar sequences, which residues are the most important in directing folding to a particular structure? Here we use molecular dynamics simulations to characterize the folding-unfolding pathways of a pair of proteins designed by Bryan and co-workers [Alexander, P. A., et al. (2005) Biochemistry 44, 14045-14054; He, Y. N., et al. (2005) Biochemistry 44, 14055-14061]. Despite being 59% identical, the two protein sequences fold to two different structures. The first sequence folds to the α+β protein G structure and the second to the all-α-helical protein A structure. We show that the final protein structure is determined early along the folding pathway. In folding to the protein G structure, the single α-helix (α1) and the β3-β4 turn fold early. Formation of the hairpin turn essentially prevents folding to helical structure in this region of the protein. This early structure is then consolidated by formation of long-range hydrophobic interactions between α and the β3-β4 turn. The protein A sequence differs both in the residues that form the β3-β4 turn and also in many of the residues that form the early hydrophobic interactions in the protein G structure. Instead, in the protein A sequence, a more hierarchical mechanism is observed, with helices folding before many of the tertiary interactions are formed. We find that small, but critical, sequence differences determine the topology of the protein early along the folding pathway, which help to explain the process by which one fold can evolve into another.
March | top
Direct Observation of Microscopic Reversibility in Single-molecule Protein Folding
Day R. and Daggett V.
Journal of Molecular Biology 366: 677-686, 2007
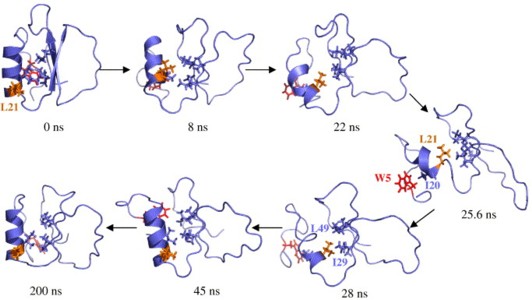
Both folded and unfolded conformations should be observed for a protein at its melting temperature (Tm), where ΔG between these states is zero. In an all-atom molecular dynamics simulation of chymotrypsin inhibitor 2 (CI2) at its experimental Tm, the protein rapidly loses its low-temperature native structure; it then unfolds before refolding to a stable, native-like conformation. The initial unfolding follows the unfolding pathway described previously for higher-temperature simulations: the hydrophobic core is disrupted, the β-sheet pulls apart and the α-helix unravels. The unfolded state reached under these conditions maintains a kernel of structure in the form of a non-native hydrophobic cluster. Refolding simply reverses this path, the side-chain interactions shift, the helix refolds, and the native packing and hydrogen bonds are recovered. The end result of this refolding is not the initial crystal structure; it contains the proper topology and the majority of the native contacts, but the structure is expanded and the contacts are long. We believe this to be the native state at elevated temperature, and the change in volume and contact lengths is consistent with experimental studies of other native proteins at elevated temperature and the chemical denaturant equivalent of Tm.
April | top
Conformational Entropy of Alanine versus Glycine in Protein Denatured States
Scott K.A., Alonso D.O.V., Sato S., Fersht A.R., and Daggett V.
Proceedings of the National Academy of Sciences USA 104: 2661-2666, 2007
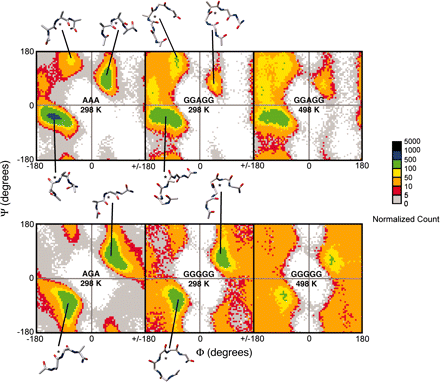
The presence of a solvent-exposed alanine residue stabilizes a helix by 0.4-2 kcal/mol relative to glycine. Various factors have been suggested to account for the differences in helical propensity, from the higher conformational freedom of glycine sequences in the unfolded state to hydrophobic and van der Waals' stabilization of the alanine side chain in the helical state. We have performed all-atom molecular dynamics simulations with explicit solvent and exhaustive sampling of model peptides to address the backbone conformational entropy difference between Ala and Gly in the denatured state. The mutation of Ala to Gly leads to an increase in conformational entropy equivalent to ≈0.4 kcal/mol in a fully flexible denatured, that is, unfolded, state. But, this energy is closely counterbalanced by the (measured) difference in free energy of transfer of the glycine and alanine side chains from the vapor phase to water so that the unfolded alanine- and glycine-containing peptides are approximately isoenergetic. The helix-stabilizing propensity of Ala relative to Gly thus mainly results from more favorable interactions of Ala in the folded helical structure. The small difference in energetics in the denatured states means that the Φ-values derived from Ala→Gly scanning of helices are a very good measure of the extent of formation of structure in proteins with little residual structure in the denatured state.
May | top
Exploring the energy landscape of protein folding using replica-exchange and conventional molecular dynamics simulations
Beck D.A.C., White G.W.N., and Daggett V.
Journal of Structural Biology 157: 514-523, 2007
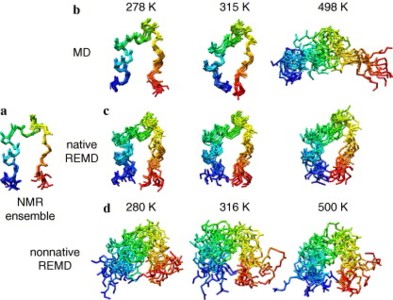
Two independent replica-exchange molecular dynamics (REMD) simulations with an explicit water model were performed of the Trp-cage mini-protein. In the first REMD simulation, the replicas started from the native conformation, while in the second they started from a nonnative conformation. Initially, the first simulation yielded results qualitatively similar to those of two previously published REMD simulations: the protein appeared to be over-stabilized, with the predicted melting temperature 50−150 K higher than the experimental value of 315 K. However, as the first REMD simulation progressed, the protein unfolded at all temperatures. In our second REMD simulation, which starts from a nonnative conformation, there was no evidence of significant folding. Transitions from the unfolded to the folded state did not occur on the timescale of these simulations, despite the expected improvement in sampling of REMD over conventional molecular dynamics (MD) simulations. The combined 1.42 μs of simulation time was insufficient for REMD simulations with different starting structures to converge. Conventional MD simulations at a range of temperatures were also performed. In contrast to REMD, the conventional MD simulations provide an estimate of Tm in good agreement with experiment. Furthermore, the conventional MD is a fraction of the cost of REMD and continuous, realistic pathways of the unfolding process at atomic resolut
October | top
The role of the turn in β-hairpin formation during WW domain folding
Sharpe T., Jonsson A.L., Rutherford T.J., Daggett V., and Fersht A.R.
Protein Science 16: 2233-2239, 2007
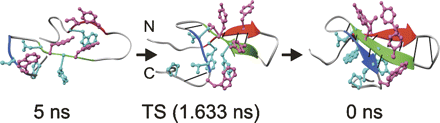
The folding of WW domains is rate limited by formation of a β-hairpin comprising residues from strands 1 and 2. Residues in the turn of this hairpin have reported Φ-values for folding close to 1 and have been proposed to nucleate folding. High Φ-values do not necessarily imply that the energetics of formation are a driving force for initiating folding. We demonstrate by NMR studies and molecular dynamics simulations that the first turn of the hYAP, FBP28, and PIN1 WW domains is structurally dynamic and solvent exposed in the native and folding transition states. It is, therefore, unlikely that the formation of the β-turn per se provides the energetic driving force for hairpin folding. It is more likely that the turn acts as an easily formed hinge that facilitates the formation of the hairpin; it is a nucleus as defined by the nucleation−condensation mechanism whereby a diffuse nucleus is stabilized by associated interactions.
November | top
Simulations of Macromolecules in Protective and Denaturing Osmolytes: Properties of Mixed Solvent Systems and Their Effects on Water and Protein Structure and Dynamics
Beck D.A.C., Bennion B.J., Alonso D.O.V., and Daggett V.
Methods in Enzymology 428: 373-396, 2007
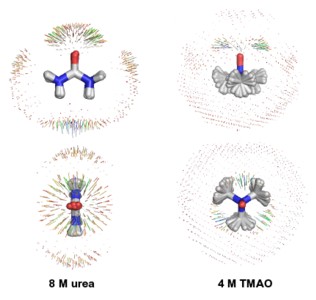
Rarely is any solution simply solute and water. In vivo, solutes, such as proteins and nucleic acids, swim in a sea of water, salts, ions, small molecules, and lipids, not to mention other macromolecules. In vitro, virtually all solutions contain a mixture of aqueous solvents, or "cosolvents" [i.e., solvent(s) in addition to water], that can alter the dynamics, behavior, solubility, and stability of proteins and nucleic acids. We have developed models for a number of cosolvents, including the denaturant urea and the small chemical chaperone trimethylamine N-oxide (TMAO). This chapter examines the models for these two cosolvents in the context of experimental data. The direct and indirect effects of these molecules on water and protein are studied with molecular dynamics simulations. These observations and conclusions are drawn from simulations of these molecules in pure water and as a cosolvent for the protein chymotrypsin inhibitor 2. Urea-induced denaturation occurs initially through attack of the protein by water and hydration of hydrophobic protein moieties as a result of disruption of the hydrogen bonding network of water by urea. This indirect denaturing effect of urea is followed by more direct action as urea replaces some waters involved in the initial hydration of the hydrophobic core and subsequently binds to polar residues and the protein main chain to compete with the intraprotein hydrogen bonds. In the case of TMAO, we find that it encourages water−water interactions, thereby stabilizing the protein as a result of the increased penalty for the hydration of hydrophobic residues.
December | top
A One-Dimensional Reaction Coordinate for Identification of Transition States from Explicit Solvent Pfold-Like Calculations
Beck D.A.C. and Daggett V.
Biophysical Journal 93: 3382-3391, 2007
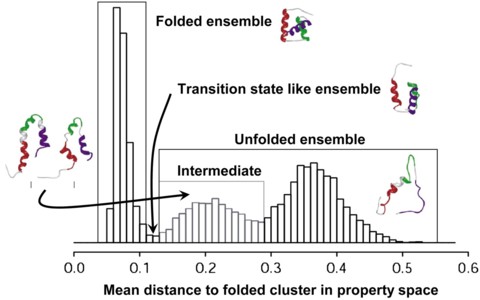
A properly identified transition state ensemble (TSE) in a molecular dynamics (MD) simulation can reveal a tremendous amount about how a protein folds and offer a point of comparison to experimentally derived ΦF values, which reflect the degree of structure in these transient states. In one such method of TSE identification, dubbed Pfold, MD simulations of individual protein structures taken from an unfolding trajectory are used to directly assess an input structure's probability of folding before unfolding, and Pfold is, by definition, 0.5 for the TSE. Other, less computationally intensive methods, such as multidimensional scaling (MDS) of the pairwise root mean-squared deviation (RMSD) matrix of the conformations sampled in a thermal unfolding trajectory, have also been used to identify the TSE. Identification of the TSE is made from the original MD simulation without the need to run further simulations. Here we present a Pfold-like study and describe methods for identification of the TSE through the derivation of a high fidelity, bounded, one-dimensional reaction coordinate for protein folding. These methods are applied to the engrailed homeodomain. The TSE identified by this approach is essentially identical to the TSE identified previously by MDS of the pairwise RMSD matrix. However, the cost of performing Pfold, or even our reduced Pfold-like calculations, is at least 36,000 times greater than the MDS method.