Paper of the Month | 2008
January | February | March | April | May | June | July | August | September | October | November | December
January | top
Conformational Changes below the Tm: Molecular Dynamics Studies of the Thermal Pretransition of Ribonuclease A
Merkley E.D., Bernard B. and Daggett V.
Biochemistry 47: 880-892, 2008
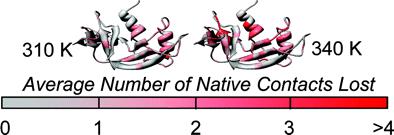
Recent work suggests that some native conformations of proteins can vary with temperature. To obtain an atomic-level description of this structural and conformational variation, we have performed all-atom, explicit-solvent molecular dynamics simulations of bovine pancreatic ribonuclease A (RNase A) up to its melting temperature (Tm ≈ 337 K). RNase A has a thermal pretransition near 320 K [Stelea, S. D., Pancoska, P., Benight, A. S., and Keiderling, T. A. (2001) Protein Sci. 10, 970-978]. Our simulations identify a conformational change that coincides with this pretransition. Between 310 and 320 K, there is a small but significant decrease in the number of native contacts, β-sheet hydrogen bonding, and deviation of backbone conformation from the starting structure, and an increase in the number of nonnative contacts. Native contacts are lost in β-sheet regions and in α1, partially due to movement of α1 away from the β-sheet core. At 330 and 340 K, a nonnative helical segment of residues 15-20 forms, corresponding to a helix observed in the N-terminal domain-swapped dimer [Liu, Y. S., Hart, P. J., Schulnegger, M. P., and Eisenberg, D. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 3437-3432]. The conformations observed at the higher temperatures possess nativelike topology and overall conformation, with many native contacts, but they have a disrupted active site. We propose that these conformations may represent the native state at elevated temperature, or the N' state. These simulations show that subtle, functionally important changes in protein conformation can occur below the Tm.
February | top
The Histamine N-Methyltransferase T105I Polymorphism Affects Active Site Structure and Dynamics
Rutherford K., Parson W.W., and Daggett V.
Biochemistry 47: 893-901, 2008
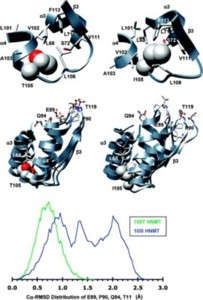
Histamine N-methyltransferase (HNMT) is the primary enzyme responsible for inactivating histamine in the mammalian brain. The human HNMT gene contains a common threonine-isoleucine polymorphism at residue 105, distal from the active site. The 105I variant has decreased activity and lower protein levels than the 105T protein. Crystal structures of both variants have been determined but reveal little regarding how the T105I polymorphism affects activity. We performed molecular dynamics simulations for both 105T and 105I at 37 °C to explore the structural and dynamic consequences of the polymorphism. The simulations indicate that replacing Thr with the larger Ile residue leads to greater burial of residue 105 and heightened intramolecular interactions between residue 105 and residues within helix α3 and strand β3. This altered, tighter packing is translated to the active site, resulting in the reorientation of several cosubstrate-binding residues. The simulations also show that the hydrophobic histamine-binding domain in both proteins undergoes a large-scale breathing motion that exposes key catalytic residues and lowers the hydrophobicity of the substrate-binding site.
March | top
Combining experiment and simulation in protein folding: closing the gap for small model systems.
Schaeffer R.D., Fersht A.R., and Daggett V.
Current Opinion in Structural Biology 18: 4-9, 2008
All-atom molecular dynamics (MD) simulations on increasingly powerful computers have been combined with experiments to characterize protein folding in detail over wider time ranges. The folding of small ultrafast folding proteins is being simulated on μs timescales, leading to improved structural predictions and folding rates. To what extent is "closing the gap" between simulation and experiment for such systems providing insights into general mechanisms of protein folding?
April | top
Different disease-causing mutations in transthyretin trigger the same conformational conversion
Steward R.E., Armen R.S., and Daggett V.
Protein Engineering Design & Selection 21: 187-195, 2008
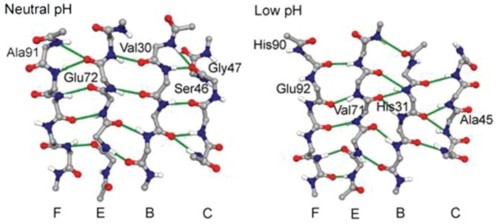
Transthyretin (TTR)-containing amyloid fibrils are deposited in cardiac tissue as a natural consequence of aging. A large number of inherited mutations lead to amyloid diseases by accelerating TTR deposition in other organs. Amyloid formation is preceded by a disruption of the quaternary structure of TTR and conformational changes in the monomer. To study conformational changes preceding the formation of amyloid, we performed molecular dynamics simulations of the wild-type monomer, amyloidogenic variants (V30M, L55P, V122I) and a protective variant (T119M) at neutral and low pH. At low pH, the D strand dissociated from the β-sheet to expose the A strand, consistent with experimental studies. In amyloidogenic variants and in the wild-type at low pH, there was a conformational change in the β-sheets into α-sheet via peptide bond flips that was not observed at neutral pH in the wild-type monomer. The same residues participated in conversion in each amyloidogenic variant simulation, originating in the G strand between residues 106 and 109, with accelerated conversion at low pH. The T119M protective variant changed the local conformation of the H strand and suppressed the conversion observed in amyloidogenic variants.
May | top
Dynameomics: mass annotation of protein dynamics and unfolding in water by high-throughput atomistic molecular dynamics simulations
Beck D.A.C, Jonsson A.L., Schaeffer R.D., Scott K.A., Day R., Toofanny R.D., Alonso D.O.V., and Daggett V.
Protein Engineering Design & Selection 21: 353-368, 2008
The goal of Dynameomics is to perform atomistic molecular dynamics (MD) simulations of representative proteins from all known folds in explicit water in their native state and along their thermal unfolding pathways. Here we present 188-fold representatives and their native state simulations and analyses. These 188 targets represent 67% of all the structures in the Protein Data Bank. The behavior of several specific targets is highlighted to illustrate general properties in the full dataset and to demonstrate the role of MD in understanding protein function and stability. As an example of what can be learned from mining the Dynameomics database, we identified a protein fold with heightened localized dynamics. In one member of this fold family, the motion affects the exposure of its phosphorylation site and acts as an entropy sink to offset another portion of the protein that is relatively immobile in order to present a consistent interface for protein docking. In another member of this family, a polymorphism in the highly mobile region leads to a host of disease phenotypes. We have constructed a web site to provide access to a novel hybrid relational/multidimensional database (described in the succeeding two papers) to view and interrogate simulations of the top 30 targets: www.dynameomics.org. The Dynameomics database, currently the largest collection of protein simulations and protein structures in the world, should also be useful for determining the rules governing protein folding and kinetic stability, which should aid in deciphering genomic information and for protein engineering and design.
June | top
Four Human Thiopurine S-Methyltransferase Alleles Severely Affect Protein Structure and Dynamics
Rutherford K. and Daggett V.
Journal of Molecular Biology 379: 803-814, 2008
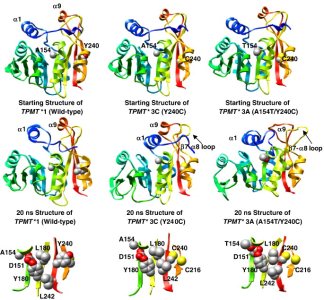
Thiopurine S-methyltransferase (TPMT) metabolizes cytotoxic thiopurine drugs used in the treatment of leukemia and inflammatory bowel disease. TPMT is a major pharmacogenomic target with 23 alleles identified to date. Several of these alleles cause rapid protein degradation and/or aggregation, making it extremely difficult to study the structural impact of the TPMT polymorphisms experimentally. We, therefore, have performed multiple molecular dynamics simulations of the four most common alleles [TPMT*2 (A80P), *3A (A154T/Y240C), *3B (A154T) and *3C (Y240C)] to investigate the molecular mechanism of TPMT inactivation at an atomic level. The A80P polymorphism in TPMT*2 disrupts helix α3 bordering the active site, which breaks several salt-bridge interactions and opens up a large cleft in the protein. The A154T polymorphism is located within the co-substrate binding site. The larger threonine alters the packing of substrate-binding residues (P68, L69, Y166), increasing the solvent exposure of the polymorphic site. This packing rearrangement may account for the complete lack of activity in the A154T mutant. The Y240C polymorphism is located in β-strand 9, distant from the active site. Side-chain contacts between residue 240 and helix α8 are lost in TPMT*3C. Residues 154 and 240 in TPMT*3A are connected through a hydrogen-bonding network. The dual polymorphisms result in a flattened, slightly distorted protein structure and an increase in the thiopurine-binding site solvent accessibility. The two variants that undergo the most rapid degradation in vivo, TPMT*2 and *3A, are also the most deformed in the simulations.
July | top
Side-chain dynamics are critical for water permeation through aquaporin-1
Smolin N., Li B., Beck D.A.C., and Daggett V.
Biophysical Journal 95: 1089-1098, 2008
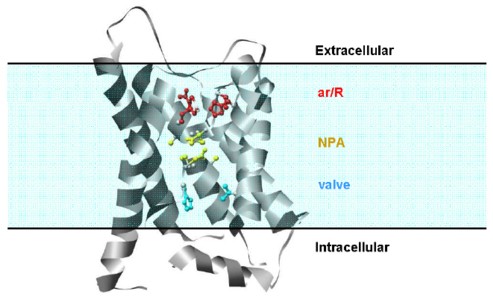
Molecular dynamics simulations of aquaporin-1 embedded in a solvated lipid bilayer were performed to investigate the mechanism of water permeation. The 2.2 Å resolution crystal structure of the bovine protein was used for five independent trajectories, lasting between 4 and 10 ns. During the equilibration and preparatory steps in which the protein was held fixed, water molecules inside the water channel adopted the same positions as observed in the crystal structure but they did not pass through the channel, suggesting that the dynamic motion of the protein is critical for water permeation. When the protein atoms were allowed to move, the side chains of the two asparagines in the two conserved NPA (Asn-Pro-Ala) motifs near the center of the channel formed hydrogen bonds with water and helped water molecules move through the channel by actively aligning them for transport. The main-chain oxygen atoms, which were exposed to the pore surface in the crystal structure, also contributed to water transfer. Besides the constriction region observed in the crystal structure (Arg 197, Phe 58, His 182 and Cys 191), we found that His 76 and Val 155 act as a valve by dynamically blocking water permeation and helping control flow.
August | top
Microscopic Reversibility of Protein Folding in Molecular Dynamics Simulations of the Engrailed Homeodomain
McCully M.E., Beck D.A.C., and Daggett V.
Biochemistry 47: 7079-7089, 2008
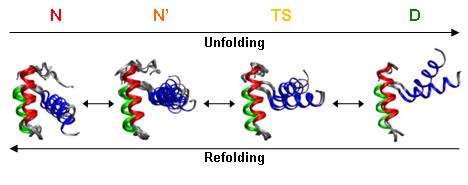
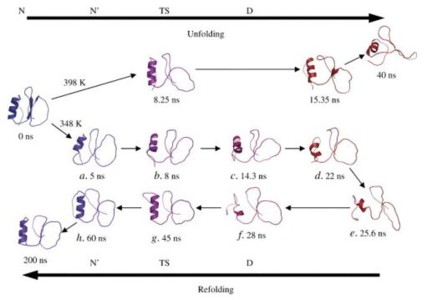
The principle of microscopic reversibility states that at equilibrium the number of molecules entering a state by a given path must equal those exiting the state via the same path under identical conditions or, in structural terms, that the conformations along the two pathways are the same. There has been some indirect evidence indicating that protein folding is such a process, but there have been few conclusive findings. In this study, we performed molecular dynamics simulations of an ultrafast unfolding and folding protein at its melting temperature to observe, on an atom-by-atom basis, the pathways the protein followed as it unfolded and folded within a continuous trajectory. In a total of 0.67 μs of simulation in water, we found six transient denaturing events near the melting temperature (323 and 330 K) and an additional refolding event following a previously identified unfolding event at a high temperature (373 K). In each case, unfolding and refolding transition state ensembles were identified, and they agreed well with experiment on the basis of a comparison of S and Φ values. On the basis of several structural properties, these 13 transition state ensembles agreed very well with each other and with four previously identified transition states from high-temperature denaturing simulations. Thus, not only were the unfolding and refolding transition states part of the same ensemble, but in five of the seven cases, the pathway the protein took as it unfolded was nearly identical to the subsequent refolding pathway. These events provide compelling evidence that protein folding is a microscopically reversible process. In the other two cases, the folding and unfolding transition states were remarkably similar to each other but the paths deviated.
September | top
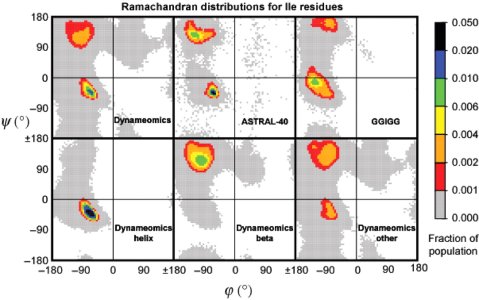
The intrinsic conformational propensities of the 20 naturally occurring amino acids and reflection of these propensities in proteins
Beck D.A.C., Alonso D.O.V., Inoyama D., and Daggett V.
Proceedings of the National Academy of Sciences USA 105: 12259-12264, 2008
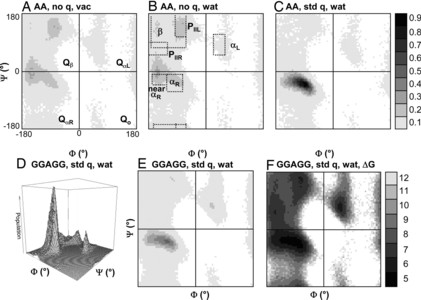
Here, we compare the distributions of main chain (Φ,Ψ) angles (i.e., Ramachandran maps) of the 20 naturally occurring amino acids in three contexts: (i) molecular dynamics (MD) simulations of Gly-Gly-X-Gly-Gly pentapeptides in water at 298 K with exhaustive sampling, where X = the amino acid in question; (ii) 188 independent protein simulations in water at 298 K from our Dynameomics Project; and (iii) static crystal and NMR structures from the Protein Data Bank. The GGXGG peptide series is often used as a model of the unstructured denatured state of proteins. The sampling in the peptide MD simulations is neither random nor uniform. Instead, individual amino acids show preferences for particular conformations, but the peptide is dynamic, and interconversion between conformers is facile. For a given amino acid, the (Φ,Ψ) distributions in the protein simulations and the Protein Data Bank are very similar and often distinct from those in the peptide simulations. Comparison between the peptide and protein simulations shows that packing constraints, solvation, and the tendency for particular amino acids to be used for specific structural motifs can overwhelm the "intrinsic propensities" of amino acids for particular (Φ,Ψ) conformations. We also compare our helical propensities with experimental consensus values using the host-guest method, which appear to be determined largely by context and not necessarily the intrinsic conformational propensities of the guest residues. These simulations represent an improved coil library free from contextual effects to better model intrinsic conformational propensities and provide a detailed view of conformations making up the "random coil" state.
October | top
Molecular Basis for the Structural Instability of Human DJ-1 Induced by the L166P Mutation Associated with Parkinson's Disease
Anderson P.C. and Daggett V.
Biochemistry 47: 9380-9393, 2008
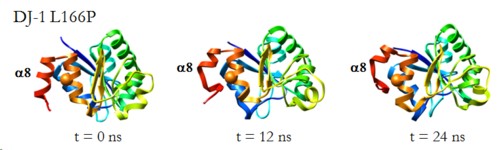
DJ-1 is a dimeric protein of unknown function in vivo. A mutation in the human DJ-1 gene causing substitution of proline for leucine at residue 166 (L166P) has been linked to early onset Parkinson's disease. Lack of structural stability has precluded experimental determination of atomic-resolution structures of the L166P DJ-1 polymorph. We have performed multiple molecular dynamics (MD) simulations (∼1/3 μs) of the wild-type and L166P DJ-1 polymorph at physiological temperature to predict specific structural effects of the L166P substitution. L166P disrupted helices α1, α5, α6 and α8 with α8 undergoing particularly severe disruption. Secondary structural elements critical for protein stability and dimerization were significantly disrupted across the entire dimer interface, as were extended hydrophobic surfaces involved in dimer formation. Relative to wild-type DJ-1, L166P DJ-1 populated a broader ensemble of structures, many of which corresponded to distorted conformations. In a L166P dimer model the substitution significantly destabilized the dimer interface, interrupting >100 intermolecular contacts that are important for dimer formation. The L166P substitution also led to major perturbations in the region of a highly conserved cysteine residue (Cys-106) that participates in dimerization and that is critical for a proposed chaperone function of DJ-1. Cys-106 is located ∼16 Å from the substitution site, demonstrating that structural disruptions propagate throughout the whole protein. Furthermore, L166P DJ-1 showed a significant increase in hydrophobic surface area relative to wild-type protein, possibly explaining the tendency of the mutant protein to aggregate. These simulations provide details about specific structural disturbances throughout L166P DJ-1 that previous studies have not revealed.
November | top
Dynameomics: Large-Scale Assessment of Native Protein Flexibility
Benson N.C. and Daggett V.
Protein Science 17: 2038-2050, 2009
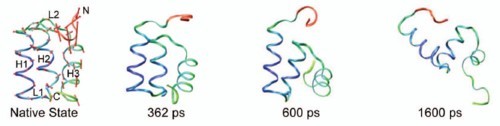
Structure is only the first step in understanding the interactions and functions of proteins. In this paper, we explore the flexibility of proteins across a broad database of over 250 solvated protein molecular dynamics simulations in water for an aggregate simulation time of ∼6 μs. These simulations are from our Dynameomics project, and these proteins represent approximately 75% of all known protein structures. We employ principal component analysis of the atomic coordinates over time to determine the primary axis and magnitude of the flexibility of each atom in a simulation. This technique gives us both a database of flexibility for many protein fold families and a compact visual representation of a particular protein's native-state conformational space, neither of which are available using experimental methods alone. These tools allow us to better understand the nature of protein motion and to describe its relationship to other structural and dynamical characteristics. In addition to reporting general properties of protein flexibility and detailing many dynamic motifs, we characterize the relationship between protein native-state flexibility and early events in thermal unfolding and show that flexibility predicts how a protein will begin to unfold. We provide evidence that fold families have conserved flexibility patterns and family members who deviate from the conserved patterns have very low sequence identity. Finally, we examine novel aspects of highly inflexible loops that are as important to structural integrity as conventional secondary structure. These loops, which are difficult if not impossible to locate without dynamic data, may constitute new structural motifs.
December | top
The V108M mutation decreases the structural stability of catechol O-methyltransferase
Rutherford K., Alphandéry E., McMillan A., Daggett V., and Parson W.W.
Biochimica et Biophysica Acta 1784: 1098-1105, 2008
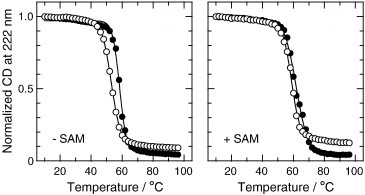
The human gene for catechol O-methyltransferase has a common single-nucleotide polymorphism that results in substitution of methionine (M) for valine (V) 108 in the soluble form of the enzyme (s-COMT). 108M s-COMT loses enzymatic activity more rapidly than 108V s-COMT at physiological temperature, and the 108M allele has been associated with increased risk of breast cancer and several neuropsychiatric disorders. We used circular dichroism (CD), dynamic light scattering, and fluorescence spectroscopy to examine how the 108V/M polymorphism affects the stability of the purified, recombinant protein to heat and guanidine hydrochloride (GuHCl). COMT contains two tryptophan residues, W143 and W38Y, which are located in loops that border the S-adenosylmethionine (SAM) and catechol binding sites. We therefore also studied the single-tryptophan mutants W38Y and W143Y in order to dissect the contributions of the individual tryptophans to the fluorescence signals. The 108V and 108M proteins differed in the stability of both the tertiary structure surrounding the active site, as probed by the fluorescence yields and emission spectra, and their global secondary structure as reflected by CD. With either probe, the midpoint of the thermal transition of 108M s-COMT was 5 to 7 °C lower than that of 108V s-COMT, and the free energy of unfolding at 25 °C was smaller by about 0.4 kcal/mol. 108M s-COMT also was more prone to aggregation or partial unfolding to a form with an increased radius of hydration at 37 °C. The co-substrate SAM stabilized the secondary structure of both 108V and 108M s-COMT. W143 dominates the tryptophan fluorescence of the folded protein and accounts for most of the decrease in fluorescence that accompanies unfolding by GuHCl. While replacing either tryptophan by tyrosine was mildly destabilizing, the lower stability of the 108M variant was retained in all cases.