Paper of the Month | 2009
January | March | June | July | August | September | October | November | December
January | top
Species variation in PrPSc protofibril models
Scouras A.D. and Daggett V.
Journal of Materials Science 43: 3625-3637, 2008
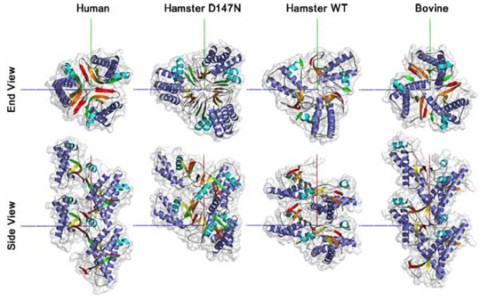
The misfolding and aggregation of the prion protein (PrP) is the primary cause of a group of infectious neurodegenerative diseases including Creutzfeldt-Jacob disease in humans and Bovine Spongiform Encephalopathy in cows. A single disease can exhibit different infectious strains distinguishable by incubation time and morphology or distribution of the aggregates. Infected brain tissue from one species can be used to infect other species, but with different efficiencies, suggesting a spectrum of species compatibility. If PrP is, as widely believed, the sole component of infection, then the species and strain differences must be accounted for by the structure of the aggregates, likely influenced by each species' PrP sequence. As there are no high-resolution data exploring this hypothesis, we performed molecular dynamics simulations of PrP for human, bovine, hamster, and D147N mutant hamster sequences at low pH to induce misfolding of the protein. We selected representative converted structures from each of the four sequences and, with the guidance of experimental data, constructed models of the infectious aggregates. Both hamster monomers showed high flexibility during conversion, suggesting hamster may more easily adopt altered conformations, which in turn may explain why it is more easily infected by some other species. Human and bovine aggregates were similar, with monomers docking in P31 symmetry to form a left-handed spiral. In contrast, hamster aggregates formed a P31 right-handed spiral. We detail the differences in the converted monomers that give rise to this difference and show that our results compare favorably with experimental data.
March | top
Characterization of cell-surface prion protein relative to its recombinant analogue: insights from molecular dynamics simulations of diglycosylated, membrane-bound human prion protein
DeMarco M.L. and Daggett V.
Journal of Neurochemistry 109: 60-73, 2009
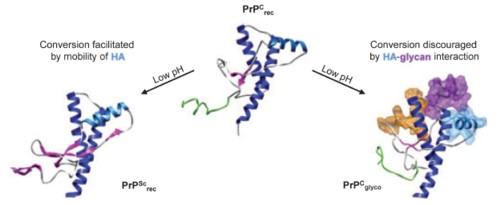
The prion protein (PrP) is responsible for several fatal neurodegenerative diseases via conversion from its normal to disease-related isoform. The recombinant form of the protein is typically studied to investigate the conversion process. This constructs lacks the co- and post-translational modifications present in vivo, there the protein has two N-linked glycans and is bound to the outer leaflet of the plasma membrane via a glycosylphosphatidylinositol (GPI) anchor. The inherent flexibility and heterogeneity of the glycans, the plasticity of the GPI anchor, and the localization of the protein in a membrane make experimental structural characterization of biological constructs of cellular prion protein (PrPC) challenging. Yet this characterization is central in determining not only the suitability of recombinant (rec)-PrPC as a model for biological forms of the protein but also the potential role of co- and post-translational modifications on the disease process. Here, we present molecular dynamics simulations of three human prion protein constructs: (i) a protein-only construct modeling the recombinant form, (ii) a diglycosylated and soluble construct, and (iii) a diglycosylated and GPI-anchored construct bound to a lipid bilayer. We found that glycosylation and membrane anchoring do not significantly alter the structure or dynamics of PrPC, but they do appreciably modify the accessibility of the polypeptide surface PrPC. In addition, the simulations of membrane-bound PrPC revealed likely recognition domains for the disease-initiating PrPC:PrPSc (infectious and/or misfolded form of the prion protein) binding event and a potential mechanism for the observed inefficiency of conversion associated with differentially glycosylated PrP species.
June | top
A Hotspot of Inactivation: The A22S and V108M Polymorphisms Individually Destabilize the Active Site Structure of Catechol O-Methyltransferase
Rutherford K.J. and Daggett V.
Biochemistry 48: 6450-6460, 2009
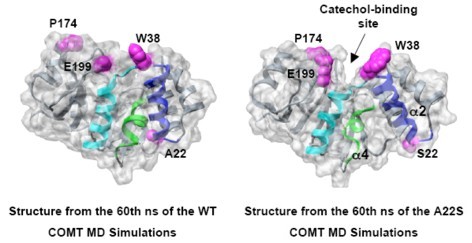
Human catechol O-methyltransferase (COMT) contains three common polymorphisms (A22S, A52T, V108M), two of which (A22S, V108M) render the protein susceptible to deactivation by temperature or oxidation. We have performed multiple molecular dynamics simulations of the wild-type, A22S, A52T, and V108M COMT proteins to explore the structural consequences of these mutations. In total, we have amassed over 1.4 μs of simulation time representing the largest set of simulations detailing the effects of polymorphisms on a protein system to date. The A52T mutation had no significant effect on COMT structure in accord with experiment, thereby serving as a good negative control for the simulation set. Residues 22 (α2) and 108 (α5) interact with each other throughout the simulations and are located in a polymorphic hotspot ∼20Å from the active site. Introduction of either the larger Ser (22) or Met (108) tightens this interaction, pulling α2 and α5 towards each other and away from the protein core. The V108M polymorphism rearranges active-site residues in α5, β3 and α6, increasing the S-adenosylmethionine site solvent exposure. The A22S mutation reorients α2, moving critical catechol-binding residues away from the substrate-binding pocket. The A22S and V108M polymorphisms evolved independently in Northern European and Asian populations. While the decreased activities of both A22S and V108M COMT are associated with an increased risk for schizophrenia, the V108M induced destabilization is also linked with improved cognitive function. These results suggest that polymorphisms within this hotspot may have evolved to regulate COMT activity and that heterozygosity for either mutation may be advantageous.
July | top
The R46Q, R131Q and R154H Polymorphs of Human DNA Glycosylase/β-Lyase hOgg1 Severely Distort the Active Site and DNA Recognition Site but do not Cause Unfolding
Anderson P.C. and Daggett V.
Journal of the American Chemical Society 131: 9506-9515, 2009
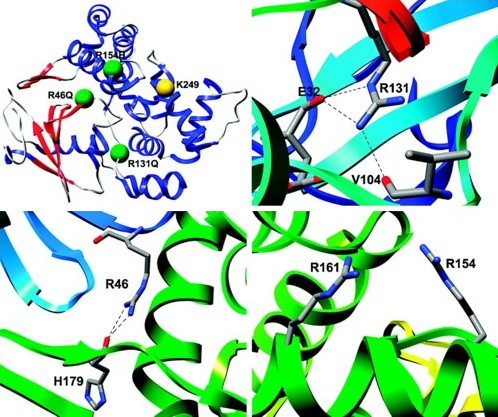
Reactive oxygen species can cause widespread cellular damage, including base alterations and strand breaks in DNA. An array of DNA-repair enzymes constitutes an essential part of the line of defense that cells use against oxidative damage to the genome. A DNA glycosylase/β-lyase enzyme, Ogg1, scavenges the genome for 8-oxoguanine, a major mutagenic DNA adduct induced by reactive oxygen species, and catalyzes its excision and subsequent cleavage of the DNA phosphate backbone. Several polymorphisms of Ogg1, including the single amino-acid substitutions R46Q, R131Q and R154H, are associated with a variety of human cancers. These three mutations have previously been characterized experimentally but no structural data have been published. We have performed multiple molecular dynamics simulations of R46Q, R131Q and R154H human Ogg1 to predict the structural and dynamical effects of the substitutions throughout the protein and specifically within the active site and substrate recognition site. None of the substitutions induced unfolding or global structural changes, instead their effects were confined principally to the active and recognition sites. Although the enzyme active site is located 18-21 Å from the three investigated mutation sites, these mutations' structural effects propagate through space and cause a major change in the orientation and chemical environment of the active site side chains. This change appears likely to compromise the ability of the Lys 249 side chain to undergo a necessary deprotonation step prior to its nucleophilic attack of the DNA. The mutations also cause an expansion of the active site cavity, which may explain the experimentally observed decreases in substrate specificity.
August | top
The consequences of pathogenic mutations to the human prion protein
Van der Kamp M.W. and Daggett V.
Protein Engineering Design & Selection 22: 461-468, 2009
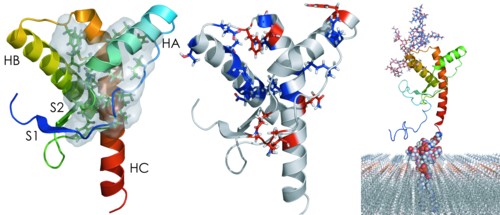
Prion diseases, in which the conformational transition of the native prion protein (PrP) to a misfolded form causes aggregation and subsequent neurodegeneration, have fascinated the scientific community as this transmissible disease appears to be purely protein-based. Disease can arise due to genetic factors only. At least 30 single point mutations have been indicated to cause disease in humans. Somehow, these mutations must influence the stability, processing and/or cellular interactions of PrP, such that aggregation can occur and disease develops. In this review, the current evidence for such effects of single point mutations is discussed, indicating that PrP can be affected in many different ways, although questions remain about the mechanism by which mutations cause disease.
September | top
Structural Changes to Monomeric CuZn Superoxide Dismutase Caused by the Familial Amyotrophic Lateral Sclerosis-Associated Mutation A4V
Schmidlin T., Kennedy B.K., and Daggett V.
Biophysical Journal 97: 1709-1718, 2009
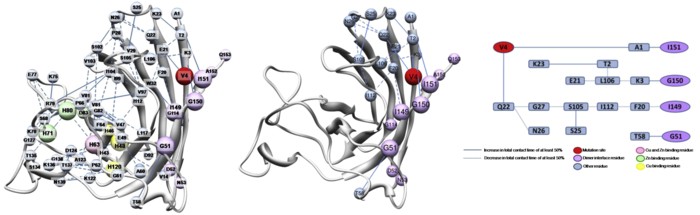
Amyotrophic lateral sclerosis (ALS) is a progressive motor neuron degenerative disease, and the inherited form, familial ALS (fALS), has been linked to over 100 different point mutations scattered throughout the Cu-Zn superoxide dismutase protein (SOD1). The disease is likely due to a toxic gain of function caused by the misfolding, oligomerization, and eventual aggregation of mutant SOD1, but it is not yet understood how the structurally diverse mutations result in a common disease phenotype. The behavior of the apo-monomer fALS-associated mutant protein A4V was explored using molecular-dynamics simulations to elucidate characteristic structural changes to the protein that may allow the mutant form to improperly associate with other monomer subunits. Simulations showed that the mutant protein is less stable than the WT protein overall, with shifts in residue-residue contacts that lead to destabilization of the dimer and metal-binding sites, and stabilization of nonnative contacts that leads to a misfolded state. These findings provide a unifying explanation for disparate experimental observations, allow a better understanding of alterations of residue contacts that accompany loss of SOD1 structural integrity, and suggest sites where compensatory changes may stabilize the mutant structure.
October | top
The V119I Polymorphism in Protein L-Isoaspartate O-Methyltransferase Alters the Substrate-Binding Interface
Rutherford K. and Daggett V.
Protein Engineering Design & Selection 22: 713-721, 2009
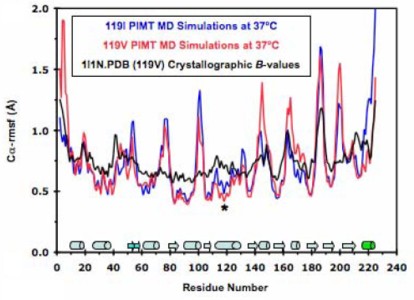
Protein L-isoaspartate O-methyltransferase (PIMT) repairs isoaspartate residues in damaged proteins, and it contains a Val-Ile polymorphismin in α5, ∼13 Å from its active site. Val119 has lower activity and thermal stability but increased affinity for endogenous substrates. Studies suggest that heterozygosity for Val/Ile favors efficient isoaspartate repair. We have performed multiple molecular dynamics simulations of 119I and 119V PIMT. Both V119 and I119 interact with the same residues throughout all of the simulations. However, the larger Ile altered the orientations of α5 and β5, both of which have co-substrate binding residues on their distal ends. I119 increases the flexibility of several residues, loosening up the S-adenosylmethionine (SAM)-binding site. These subtle changes are propagated towards the isoaspartate-docking site via residues common to both active sites. The increased mobility in 119I PIMT reorients α3, resulting in a salt-bridge network at the substrate-binding interface that disrupts several key side-chain interactions in the isoaspartate site. In contrast, 119V PIMT remains quite rigid with little change to the co-substrate binding site, which could hinder SAM's binding and release, accounting for the decreased activity. These results shed light on the molecular basis behind the decreased activity and increased specificity for endogenous substrates of 119V PIMT relative to the 119I variant. 119I PIMT catalyzes the methylation reaction but may have difficulties recognizing and orienting specific substrates due to its distorted substrate-binding site. Heterozygosity for both the Ile and Val alleles may provide the best of both worlds, allowing the fast and specific methylation of damaged proteins.
November | top
Principles of Ligand Binding within a Completely Buried Cavity in HIF2α PAS-B
Key J., Scheuermann T.H., Anderson P.C., Daggett V., and Gardner K.H.
Journal of the American Chemical Society 131: 17647-17654, 2009
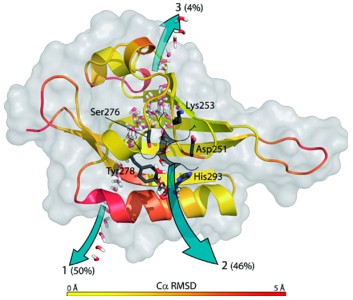
Hypoxia-inducible factors (HIFs) are heterodimeric transcription factors responsible for the metazoan hypoxia response and promote tumor growth, metastasis, and resistance to cancer treatment. The C-terminal Per-ARNT-Sim (PAS) domain of HIF2α (HIF2a PAS-B) contains a preformed solvent-inaccessible cavity that binds artificial ligands that allosterically perturb the formation of the HIF heterodimer. To better understand how small molecules bind within this domain, we examined the structures and equilibrium and transition-state thermodynamics of HIF2α PAS-B with several artificial ligands using isothermal titration calorimetry, NMR exchange spectroscopy, and X-ray crystallography. Rapid association rates reveal that ligand binding is not dependent upon a slow conformational change in the protein to permit ligand access, despite the closed conformation observed in the NMR and crystal structures. Compensating enthalpic and entropic contributions to the thermodynamic barrier for ligand binding suggest a binding-competent transition state characterized by increased structural disorder. Finally, molecular dynamics simulations reveal conversion between open and closed conformations of the protein and pathways of ligand entry into the binding pocket.
December | top
Dynameomics: A Consensus View of the Protein Unfolding/Folding Transition State Ensemble across a Diverse Set of Protein Folds
Jonsson A.L., Scott K.A., and Daggett V.
Biophysical Journal 97: 2958-2966, 2009
The Dynameomics project aims to simulate a representative sample of all globular protein metafolds under both native and unfolding conditions. We have identified protein unfolding transition state (TS) ensembles from multiple molecular dynamics simulations of high-temperature unfolding in 183 structurally distinct proteins. These data can be used to study individual proteins and individual protein metafolds and to mine for TS structural features common across all proteins. Separating the TS structures into four different fold classes (all proteins, all-α, all-β, and mixed α/β and α + β) resulted in no significant difference in the overall protein properties. The residues with the most contacts in the native state lost the most contacts in the TS ensemble. On average, residues beginning in an a-helix maintained more structure in the TS ensemble than did residues starting in β-strands or any other conformation. The metafolds studied here represent 67% of all known protein structures, and this is, to our knowledge, the largest, most comprehensive study of the protein folding/unfolding TS ensemble to date. One might have expected broad distributions in the average global properties of the TS relative to the native state, indicating variability in the amount of structure present in the TS. Instead, the average global properties converged with low standard deviations across metafolds, suggesting that there are general rules governing the structure and properties of the TS.