Paper of the Month | 2010
January | February | April | June | July | September | November | December
January | top
The relationship between water bridges and the polyproline II conformation: a large-scale analysis of molecular dynamics simulations and crystal structures
Law P.B. and Daggett V.
Proten Engineering Design & Selection 23: 27-33, 2010
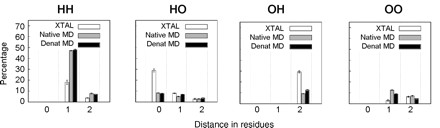
It has been suggested that denatured proteins are predisposed toward the left-handed polyproline II (PII) conformation. One possible source of PII stability in the denatured state is water bridges. Water bridges are networks of water molecules that link nearby hydrogen bond acceptors and/or donors on proteins. On the basis of the proposed behavior of PII and water bridges, the propensity of a residue to participate in water bridges should be correlated with its PII propensity. To test this hypothesis, we analyzed the following data sets: 2351 high-resolution crystal structures, and the native and denatured states of 188 different proteins from all-atom, explicit-solvent molecular dynamics (MD) simulations, which are part of our Dynameomics effort. We found that water bridges do not explain the high frequency of PII in denatured states; such bridges are less frequent around PII than around other conformations. Thus, this analysis casts doubt on water bridges as a dominant factor determining the residue-based PII propensities.
February | top
Temperature dependence of the flexibility of thermophilic and mesophilic flavoenzymes of the nitroreductase fold
Merkley E.D., Parson W.W., and Daggett V.
Proten Engineering Design & Selection 23:327-336, 2010
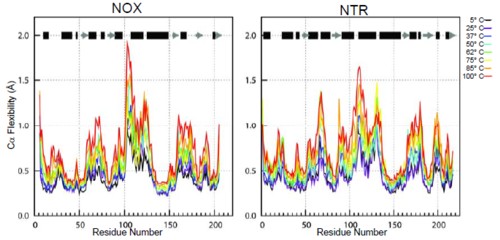
A widely held hypothesis regarding the thermostability of thermophilic proteins states asserts that, at any given temperature, thermophilic proteins are more rigid than their mesophilic counterparts. Many experimental and computational studies have addressed this question with conflicting results. Here, we compare two homologous enzymes, one mesophilic (Escherichia coli FMN-dependent nitroreductase; NTR) and one thermophilic (Thermus thermophilus NADH oxidase; NOX), by multiple molecular dynamics simulations at temperatures from 5 to 100°C. We find that the global rigidity/flexibility of the two proteins, assessed by a variety of metrics, is similar on the time scale of our simulations. However, the thermophilic enzyme retains its native conformation to a much greater degree at high temperature than does the mesophilic enzyme, both globally and within the active site. The simulations identify the helix F-helix G 'arm' as the region with the greatest difference in loss of native contacts between the two proteins with increasing temperature. In particular, a network of electrostatic interactions holds helix F to the body of the protein in the thermophilic protein, and this network is absent in the mesophilic counterpart.
April | top
Dynameomics: A Comprehensive Database of Protein Dynamics
Van der Kamp M.W., Schaeffer R.D., Jonsson A.L., Scouras A.D., Simms A.M., Toofanny R.D., Benson N.C., Anderson P.C., Merkley E.D., Rysavy S., Bromley D., Beck D.A.C., and Daggett V.
Structure 18:423-435, 2010

The dynamic behavior of proteins is important for an understanding of their function and folding. We have performed molecular dynamics simulations of the native state and unfolding pathways of over 2000 protein/peptide systems (∼11,000 independent simulations) representing the majority of folds in globular proteins. These data are stored and organized using an innovative database approach, which can be mined to obtain both general and specific information about the dynamics and folding/unfolding of proteins, relevant subsets thereof, and individual proteins. Here we describe the project in general terms and the type of information contained in the database. Then we provide examples of mining the database for information relevant to protein folding, structure building, the effect of single-nucleotide polymorphisms, and drug design. The native state simulation data and corresponding analyses for the 100 most populated metafolds, together with related resources, are publicly accessible through www.dynameomics.org.
June | top
Polymorphisms and disease: hotspots of inactivation in methyltransferases
Rutherford K. and Daggett V.
Trends in Biochemical Sciences 35:531-538, 2010
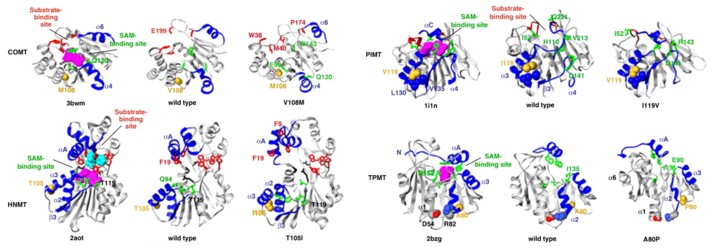
Methyltransferases catalyze the methylation processes essential for protein/DNA repair, transcriptional regulation, and drug metabolism in vivo. More than 500 human methyltransferase polymorphisms have been identified, many of which are linked to disease. We mapped all available coding polymorphisms of seven methyltransferases onto their structures to address their structural significance, and identified a polymorphic hotspot not, vert, ∼20 Å from the active site in four of the proteins. Molecular dynamics simulations of these proteins reveal a common mechanism of destabilization: the mutations alter important side-chain contacts within the polymorphic site that are propagated through the protein, thereby distorting the active site. We propose that this hotspot might have arisen to modulate enzymatic activity, with decreased activity actually conferring an advantage in three of the four methyltransferases.
July | top
A Comprehensive Multidimensional-Embedded, One-Dimensional Reaction Coordinate for Protein Unfolding/Folding
Toofanny R.D., Jonsson A.L., and Daggett V.
Biophysical Journal 98:2671-2681, 2010
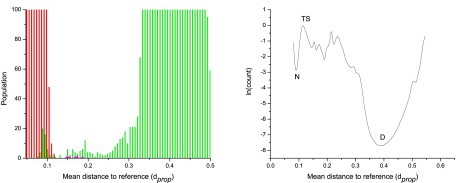
The goal of the Dynameomics project is to perform, store, and analyze molecular dynamics simulations of representative proteins, of all known globular folds, in their native state and along their unfolding pathways. To analyze unfolding simulations, the location of the protein along the unfolding reaction coordinate (RXN) must be determined. Properties such as the fraction of native contacts and radius of gyration are often used; however, there is an issue regarding degeneracy with these properties, as native and nonnative species can overlap. Here, we used 15 physical properties of the protein to construct a multidimensional-embedded, one-dimensional RXN coordinate that faithfully captures the complex nature of unfolding. The unfolding RXN coordinates for 188 proteins (1534 simulations and 22.9 μs in explicit water) were calculated. Native, transition, intermediate, and denatured states were readily identified with the use of this RXN coordinate. A global native ensemble based on the native-state properties of the 188 proteins was created. This ensemble was shown to be effective for calculating RXN coordinates for folds outside the initial 188 targets. These RXN coordinates enable, high-throughput assignment of conformational states, which represents an important step in comparing protein properties across fold space as well as characterizing the unfolding of individual proteins.
September | top
Refolding the Engrailed Homeodomain: Structural Basis for the Accumulation of a Folding Intermediate
McCully M.E., Beck D.A.C., Fersht A.R., and Daggett V.
Biophysical Journal 99:1628-1636, 2010
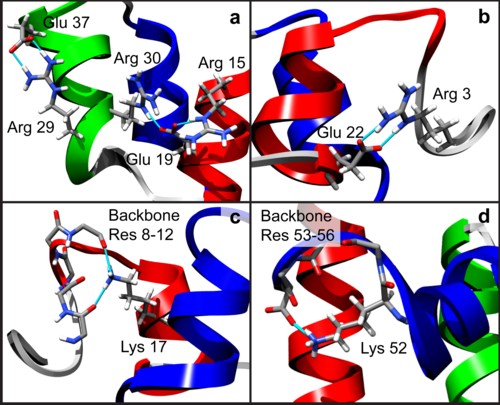
The ultrafast folding pathway of the engrailed homeodomain has been exceptionally well characterized by experiment and simulation. Helices II and III of the three-helix bundle protein form the native helix-turn-helix motif as an on-pathway intermediate within a few microseconds. The slow step is then the proper docking of the helices in ∼15 μs. However, there is still the unexplained puzzle of why helix docking is relatively slow, which is part of the more general question as to why rearrangements of intermediates occur slowly. To address this problem, we performed 46 all-atom molecular dynamics refolding simulations in explicit water, for a total of 15 μs of simulation time. The simulations started from an intermediate state structure that was generated in an unfolding simulation at 498 K and was then quenched to folding-permissive temperatures. The protein refolded successfully in only one of the 46 simulations, and in that case the refolding pathway mirrored the unfolding pathway at high temperature. In the 45 simulations in which the protein did not fully fold, nonnative salt bridges trapped the protein, which explains why the protein folds relatively slowly from the intermediate state.
November | top
The influence of pH on the human prion protein: Insights into the early steps of misfolding
Van der Kamp M.W. and Daggett V.
Biophysical Journal 99:2289-2298, 2010
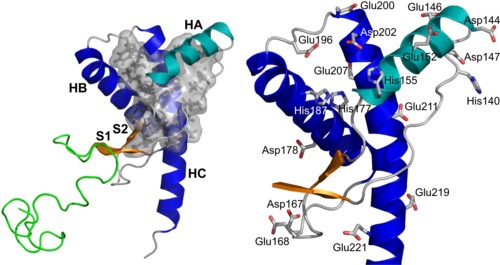
Transmissible spongiform encephalopathies, or prion diseases, are caused by misfolding and aggregation of the prion protein PrP. Conversion from the normal cellular form (PrPC) or recombinant PrP (recPrP) to a misfolded form is pH-sensitive, in that misfolding and aggregation occur more readily at lower pH. To gain more insight into the influence of pH on the dynamics of PrP and its potential to misfold, we performed extensive molecular-dynamics simulations of the recombinant PrP protein (residues 90-230) in water at three different pH regimes: neutral (or cytoplasmic) pH (∼7.4), middle (or endosomal) pH (∼5), and low pH (<4). We present five different simulations of 50 ns each for each pH regime, amounting to a total of 750 ns of simulation time. A detailed analysis and comparison with experiment validate the simulations and lead to new insights into the mechanism of pH-induced misfolding. The mobility of the globular domain increases with decreasing pH, through displacement of the first helix and instability of the hydrophobic core. At middle pH, conversion to a misfolded (PrPSc-like) conformation is observed. The observed changes in conformation and stability are consistent with experimental data and thus provide a molecular basis for the initial steps in the misfolding process.
December | top
Diverse Effects on the Native β-Sheet of the Human Prion Protein Due to Disease-Associated Mutations
Chen W., Van der Kamp M.W., and Daggett V.
Biochemistry 49:9874-9881, 2010
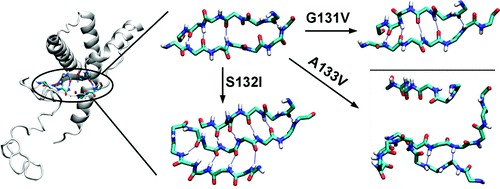
Prion diseases are fatal neurodegenerative disorders that involve the conversion of the normal cellular form of the prion protein (PrPC) to a misfolded pathogenic form (PrPSc). There are many genetic mutations of PrP associated with human prion diseases. Three of these point mutations are located at the first strand of the native β-sheet in human PrP: G131V, S132I, and A133V. To understand the underlying structural and dynamic effects of these disease-causing mutations on the human PrP, we performed molecular dynamics of wild-type and mutated human PrP. The results indicate that the mutations induced different effects but they were all related to misfolding of the native β-sheet: G131V caused the elongation of the native β-sheet, A133V disrupted the native β-sheet, and S132I converted the native β-sheet to an α-sheet. The observed changes were due to the reorientation of side chain-side chain interactions upon introducing the mutations. In addition, all mutations impaired a structurally conserved water site at the native β-sheet. Our work suggests various misfolding pathways for human PrP in response to mutation.