Paper of the Month | 2011
January | February | March | May | June | July | August | October | November
January | top
Generation of a consensus protein domain dictionary
Schaeffer R.D., Jonsson A.L., Simms A.M., and Daggett V.
Bioinformatics 27: 46-54, 2011
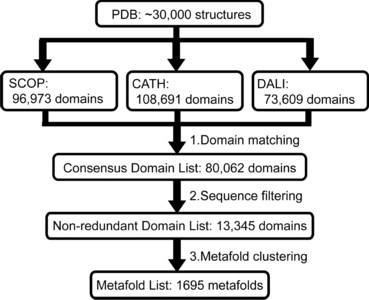
Motivation: The discovery of new protein folds is a relatively rare occurrence even as the rate of protein structure determination increases. This rarity reinforces the concept of folds as reusable units of structure and function shared by diverse proteins. If the folding mechanism of proteins is largely determined by their topology, then the folding pathways of members of existing folds could encompass the full set used by globular protein domains.
Results: We have used recent versions of three common protein domain dictionaries (SCOP, CATH and Dali) to generate a consensus domain dictionary (CDD). Surprisingly, 40% of the metafolds in the CDD are not composed of autonomous structural domains, i.e. they are not plausible independent folding units. This finding has serious ramifications for bioinformatics studies mining these domain dictionaries for globular protein properties. However, our main purpose in deriving this CDD was to generate an updated CDD to choose targets for MD simulation as part of our dynameomics effort, which aims to simulate the native and unfolding pathways of representatives of all globular protein consensus folds (metafolds). Consequently, we also compiled a list of representative protein targets of each metafold in the CDD.
Availability and implementation: This domain dictionary is available at www.dynameomics.org.
February | top
The dynameomics rotamer library: Amino acid side chain conformations and dynamics from comprehensive molecular dynamics simulations in water
Scouras A.D. and Daggett V.
Protein Science 20: 341-352, 2011
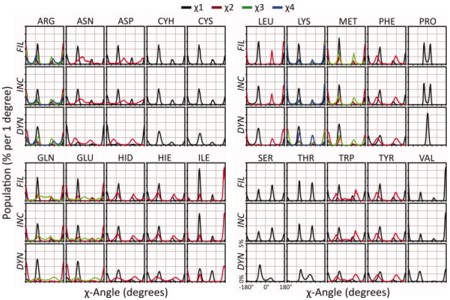
We have recently completed systematic molecular dynamics simulations of 807 different proteins representing 95% of the known autonomous protein folds in an effort we refer to as Dynameomics. Here we focus on the analysis of side chain conformations and dynamics to create a dynamic rotamer library. Overall this library is derived from 31,000 occurrences of each of 86,217 different residues, or 2.7 x 109 rotamers. This dynamic library has 74% overlap of rotamer distributions with rotamer libraries derived from static high-resolution crystal structures. Seventy-five percent of the residues had an assignable primary conformation, and 68% of the residues had at least one significant alternate conformation. The average correlation time for switching between rotamers ranged from 22 ps for Met to over 8 ns for Cys; this time decreased 20-fold on the surface of the protein and modestly for dihedral angles further from the main chain. Side chain S2 axis order parameters were calculated and they correlated well with those derived from NMR relaxation experiments (R = 0.9). Relationships relating the S2 axis order parameters to rotamer occupancy were derived. Overall the Dynameomics rotamer library offers a comprehensive depiction of side chain rotamer preferences and dynamics in solution, and more realistic distributions for dynamic proteins in solution at ambient temperature than libraries derived from crystal structures, in particular charged surface residues are better represented. Details of the rotamer library are presented here and the library itself can be downloaded at http://www.dynameomics.org.
March | top
The denatured state dictates the topology of two proteins with almost identical sequence but different native structure and function
Morronea A., McCully M.E., Bryan P.N., Brunori M., Daggett V., Gianni S., Travaglini-Allocatelli C.
The Journal of Biological Chemistry 286: 3863-3872, 2011
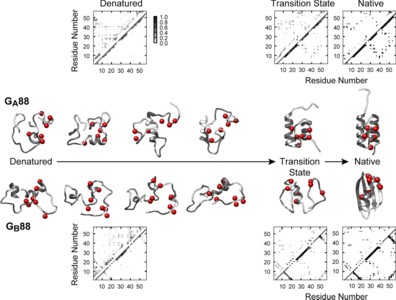
The protein folding problem is often studied by comparing the mechanisms of proteins sharing the same structure but different sequence. The recent design of the two proteins GA88 and GB88, displaying different structures and functions while sharing 88% sequence identity (49 out of 56 amino acids), allows the unique opportunity for a complementary approach. At which stage of its folding pathway does a protein commit to a given topology? Which residues are crucial in directing folding mechanisms to a given structure? By using a combination of biophysical and computational techniques, we have characterized the folding of both GA88 and GB88. We show that, contrary to expectation, GB88, characterized by a native α+β fold, displays in the denatured state a content of native-like helical structure greater than GA88, which is all-α in its native state. Both experiments and simulations indicate that such residual structure may be tuned by changing pH. Thus, despite the high sequence identity, the folding pathways for these two proteins appear to diverge as early as in the denatured state. Our results suggest a mechanism whereby protein topology is committed very early along the folding pathway, being imprinted in the residual structure of the denatured state.
May | top
Manifestations of Native Topology in the Denatured State Ensemble of Rhodopseudomonas palustris Cytochrome c′
Dar T.A., Schaeffer R.D., Daggett V., and Bowler B.E.
Biochemistry 50:1029-1041, 2011
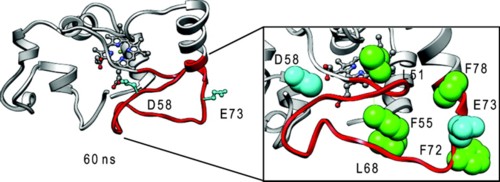
To provide insight into the role of local sequence in the nonrandom coil behavior of the denatured state, we have extended our measurements of histidine-heme loop formation equilibria for cytochrome c′ to 6 M guanidine hydrochloride. We observe that there is some reduction in the scatter about the best fit line of loop stability versus loop size data in 6 M versus 3 M guanidine hydrochloride, but the scatter is not eliminated. The scaling exponent, ν3, of 2.5 ± 0.2 is also similar to that found previously in 3 M guanidine hydrochloride (2.6 ± 0.3). Rates of histidine-heme loop breakage in the denatured state of cytochrome c′ show that some histidine-heme loops are significantly more persistent than others at both 3 and 6 M guanidine hydrochloride. Rates of histidine-heme loop formation more closely approximate random coil behavior. This observation indicates that heterogeneity in the denatured state ensemble results mainly from contact persistence. When mapped onto the structure of cytochrome c′, the histidine-heme loops with slow breakage rates coincide with chain reversals between helices 1 and 2 and between helices 2 and 3. Molecular dynamics simulations of the unfolding of cytochrome c′ at 498 K show that these reverse turns persist in the unfolded state. Thus, these portions of the primary structure of cytochrome c′ set up the topology of cytochrome c′ in the denatured state, predisposing the protein to fold efficiently to its native structure.
June | top
Structural Effects of the L145Q, V157F, and R282W Cancer-Associated Mutations in the p53 DNA-Binding Core Domain
Calhoun S. and Daggett V.
Biochemistry 50:5345-5353, 2011
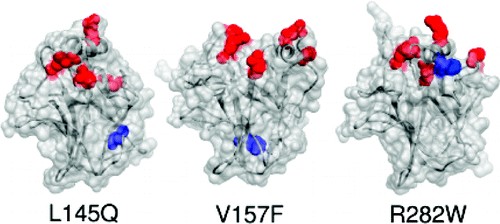
The p53 tumor suppressor is a transcription factor involved in many important signaling pathways, such as apoptosis and cell-cycle arrest. In over half of human cancers, p53 function is compromised by a mutation in its gene. Mutations in the p53 DNA-binding core domain destabilize the structure and reduce DNA-binding activity. We performed molecular dynamics simulations at physiological temperature to study the structural and dynamic effects of the L145Q, V157F, and R282W cancer-associated mutations in comparison to the wild-type protein. While there were common regions of destabilization in the mutant simulations, structural changes particular to individual mutations were also observed. Significant backbone deviations of the H2 helix and S7-S8 loop were observed in all mutant simulations; the H2 helix binds to DNA. In addition, the L145Q and V157F mutations, which are located in the β-sandwich core of the domain, disrupted the β-sheet structure and the loop-sheet-helix motif. The R282W mutation caused distortion of the loop-sheet-helix motif, but otherwise this mutant was similar to the wild-type structure. The introduction of these mutations caused rearrangement of the DNA-binding surface, consistent with their reduced DNA-binding activity. The simulations reveal detailed effects of the mutations on the stability and dynamics of p53 that may provide insight for therapeutic approaches.
July | top
Malleability of folding intermediates in the homeodomain superfamily
Banachewicz W., Religa T.L., Schaeffer R.D., Daggett V., and Fersht A.R.
Proceedings of the National Academy of Sciences USA 108:5596-5601, 2011
Members of the homeodomain superfamily are three-helix bundle proteins whose second and third helices form a helix-turn-helix motif (HTH). Their folding mechanism slides from the ultrafast, three-state framework mechanism for the engrailed homeodomain (EnHD), in which the HTH motif is independently stable, to an apparent two-state nucleation-condensation model for family members with an unstable HTH motif. The folding intermediate of EnHD has nearly native HTH structure, but it is not docked with helix1. The determinant of whether two- or three-state folding was hypothesized to be the stability of the HTH substructure. Here, we describe a detailed Φ-value analysis of the folding of the Pit1 homeodomain, which has similar ultrafast kinetics to that of EnHD. Formation of helix1 was strongly coupled with formation of HTH, which was initially surprising because they are uncoupled in the EnHD folding intermediate. However, we found a key difference between Pit1 and EnHD: The isolated peptide corresponding to the HTH motif in Pit1 was not folded in the absence of H1. Independent molecular dynamics simulations of Pit1 unfolding found an intermediate with H1 misfolded onto the HTH motif. The Pit1 folding pathway is the connection between that of EnHD and the slower folding homeodomains and provides a link in the transition of mechanisms from two- to three-state folding in this superfamily. The malleability of folding intermediates can lead to unstable substructures being stabilized by a variety of nonnative interactions, adding to the continuum of folding mechanisms.
August | top
Implementation of 3D spatial indexing and compression in a large-scale molecular dynamics simulation database for rapid atomic contact detection
Toofanny R.D., Simms A.M., Beck D.A.C., and Daggett V.
BMC Bioinformatics 12:334, 2011

Background
Molecular dynamics (MD) simulations offer the ability to observe the dynamics and interactions of both whole macromolecules and individual atoms as a function of time. Taken in context with experimental data, atomic interactions from simulation
provide insight into the mechanics of protein folding, dynamics, and function. The calculation of atomic interactions or contacts from an MD trajectory is computationally demanding and the work required grows exponentially with the size
of the simulation system. We describe the implementation of a spatial indexing algorithm in our multi-terabyte MD simulation database that significantly reduces the run-time required for discovery of contacts. The approach is applied to
the Dynameomics project data. Spatial indexing, also known as spatial hashing, is a method that divides the simulation space into regular sized bins and attributes an index to each bin. Since, the calculation of contacts is widely employed
in the simulation field, we also use this as the basis for testing compression of data tables. We investigate the effects of compression of the trajectory coordinate tables with different options of data and index compression within MS
SQL SERVER 2008.
Results
Our implementation of spatial indexing speeds up the calculation of contacts over a 1 nanosecond (ns) simulation window by between 14% and 90% (i.e., 1.2 and 10.3 times faster). For a 'full' simulation trajectory (51 ns) spatial indexing
reduces the calculation run-time between 31 and 81% (between 1.4 and 5.3 times faster). Compression resulted in reduced table sizes but resulted in no significant difference in the total execution time for neighbour discovery. The greatest
compression (~36%) was achieved using page level compression on both the data and indexes.
Conclusions
The spatial indexing scheme significantly decreases the time taken to calculate atomic contacts and could be applied to other multidimensional neighbor discovery problems. The speed up enables on-the-fly calculation and visualization of
contacts and rapid cross simulation analysis for knowledge discovery. Using page compression for the atomic coordinate tables and indexes saves ~36% of disk space without any significant decrease in calculation time and should be considered
for other non-transactional databases in MS SQL SERVER 2008.
October | top
The effect of context on the folding of β-hairpins
Jonsson A.L. and Daggett V.
Journal of Structural Biology 176:143-150, 2011
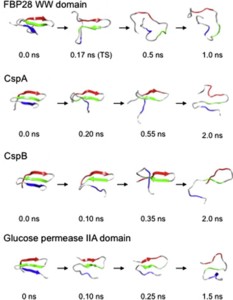
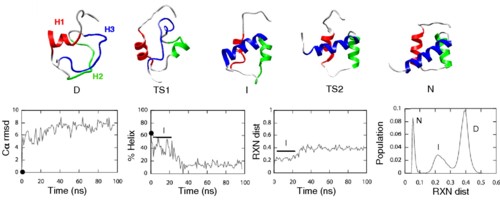
Small β-hairpin peptides have been widely used as models for the folding of β-sheets. But how applicable is the folding of such models to β-structure in larger proteins with conventional hydrophobic cores? Here we present multiple unfolding simulations of three such proteins that contain the WW domain double hairpin β-sheet motif: cold shock protein A (CspA), cold shock protein B (CspB) and glucose permease IIA domain. We compare the behavior of the free motif in solution and in the context of proteins of different size and architecture. Both Csp proteins lost contacts between the double-hairpin motif and the protein core as the first step of unfolding and proceeded to unfold with loss of the third β-strand, similar to the isolated WW domain. The glucose permease IIA domain is a larger protein and the contacts between the motif and the core were not lost as quickly. Instead the unfolding pathway of glucose permease IIA followed a different pathway with β1 pulling away from the sheet first. Interestingly, when the double hairpin motif was excised from the glucose permease IIA domain and simulated in isolation in water it unfolded by the same pathway as the WW domain, indicating that it is tertiary interactions with the protein that alter the motif's unfolding not a sequence dependent effect on its intrinsic unfolding behavior. With respect to the unfolding of the hairpins, there was no consistent order to the loss of hydrogen bonds between the β-strands in the hairpins in any of the systems. Our results show that while the folding behavior of the isolated WW domain is generally consistent with the double hairpin motif's behavior in the cold shock proteins, it is not the case for the glucose permease IIA domain. So, one must be cautious in extrapolating findings from model systems to larger more complicated proteins where tertiary interactions can overwhelm intrinsic behavior.
November | top
Protein simulation data in the relational model
Simms A.M. and Daggett V.
The Journal of Supercomputing, In Press, 2011
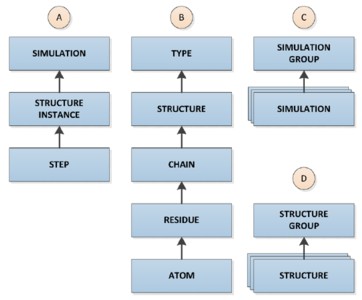
High performance computing is leading to unprecedented volumes of data. Relational databases offer a robust and scalable model for storing and analyzing scientific data. However, these features do not come without a cost--significant design effort is required to build a functional and efficient repository. Modeling protein simulation data in a relational database presents several challenges: The data captured from individual simulations are large, multidimensional, and must integrate with both simulation software and external data sites. Here, we present the dimensional design and relational implementation of a comprehensive data warehouse for storing and analyzing molecular dynamics simulations using SQL Server.
December | top
A temperature-dependent conformational change of NADH oxidase from Thermus thermophilus HB8
Merkley E.D., Daggett V., and Parson W.W.
Proteins: Structure, Function, and Bioinformatics 80:546-555, 2012
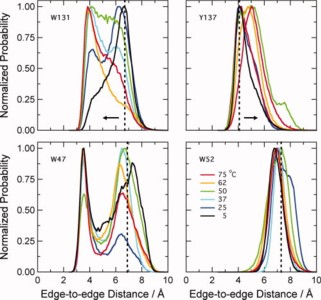
Using molecular dynamics simulations and steady-state fluorescence spectroscopy, we have identified a conformational change in the active site of a thermophilic flavoenzyme, NADH oxidase from Thermus thermophilus HB8 (NOX). The enzyme's far-UV circular dichroism spectrum, intrinsic tryptophan fluorescence, and apparent molecular weight measured by dynamic light scattering varied little between 25 and 75°C. However, the fluorescence of the tightly bound FAD cofactor increased approximately fourfold over this temperature range. This effect appears not to be due to aggregation, unfolding, cofactor dissociation, or changes in quaternary structure. We therefore attribute the change in flavin fluorescence to a temperature-dependent conformational change involving the NOX active site. Molecular dynamics simulations and the effects of mutating aromatic residues near the flavin suggest that the change in fluorescence results from a decrease in quenching by electron transfer from tyrosine 137 to the flavin.