Paper of the Month | 2012
January | April | June | July | August | September
January | top
GB1 Is Not a Two-State Folder: Identification and Characterization of an On-Pathway Intermediate
Morrone A., Giri R., Toofanny R.D., Travaglini-Allocatelli C., Brunori M., Daggett V., and Gianni S.
Biophysical Journal 101: 2053-2060, 2012
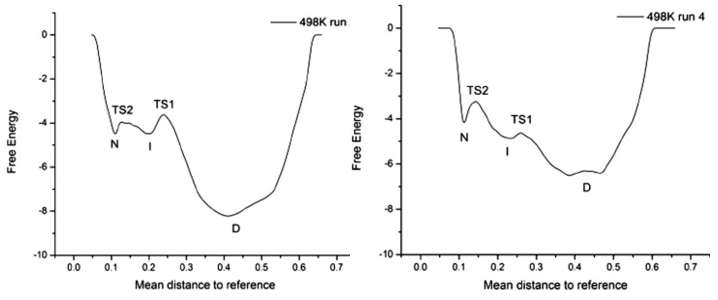
The folding pathway of the small α/β protein GB1 has been extensively studied during the past two decades using both theoretical and experimental approaches. These studies provided a consensus view that the protein folds in a two-state manner. Here, we reassessed the folding of GB1, both by experiments and simulations, and detected the presence of an on-pathway intermediate. This intermediate has eluded earlier experimental characterization and is distinct from the collapsed state previously identified using ultrarapid mixing. Failure to identify the presence of an intermediate affects some of the conclusions that have been drawn for GB1, a popular model for protein folding studies.
April | top
A temperature-dependent conformational change of NADH oxidase from Thermus thermophilus HB8
Merkley E.D., Daggett V., and Parson W.W.
Proteins: Structure, Function, and Bioinformatics 80: 546-555, 2012
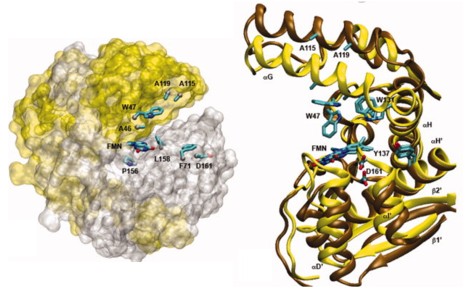
Using molecular dynamics simulations and steady-state fluorescence spectroscopy, we have identified a conformational change in the active site of a thermophilic flavoenzyme, NADH oxidase from Thermus thermophilus HB8 (NOX). The enzyme's far-UV circular dichroism spectrum, intrinsic tryptophan fluorescence, and apparent molecular weight measured by dynamic light scattering varied little between 25 and 75°C. However, the fluorescence of the tightly bound FAD cofactor increased approximately fourfold over this temperature range. This effect appears not to be due to aggregation, unfolding, cofactor dissociation, or changes in quaternary structure. We therefore attribute the change in flavin fluorescence to a temperature-dependent conformational change involving the NOX active site. Molecular dynamics simulations and the effects of mutating aromatic residues near the flavin suggest that the change in fluorescence results from a decrease in quenching by electron transfer from tyrosine 137 to the flavin.
June | top
Disruption of the X-loop turn of the prion protein linked to scrapie resistance
Scouras A.D. and Daggett V.
Protein Engineering, Design and Selection 25: 243-249, 2012
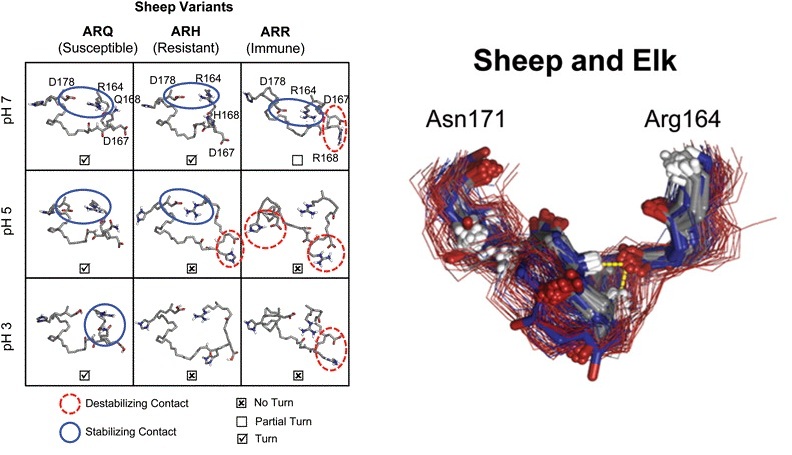
The prion diseases are a class of neurodegenerative diseases caused by the misfolding and aggregation of the prion protein (PrPC) into toxic and infectious oligomers (PrPSc). These oligomers are critical to understanding and combating these diseases. Differences in the sequence of PrP affect disease susceptibility, likely by shifting the tolerance of the protein for adaptation to PrPSc conformations and/or the recognition event between PrPSc and PrP C prior to conversion of the PrPC. We selected two sets of PrPSc-resistant mutant sequences for solvated atomistic molecular dynamics simulation to investigate the structural basis of resistance. The first group involved mutation in the X-loop (residues 164-171) resulting from selective breeding of sheep. The second group included eight mutants in mice identified by random mutagenesis targeting helix C followed by screening in cell cultures. Multiple simulations were performed of 14 different mutant and control constructs under different pH conditions for a total of 3.6 .s of simulation time. The X-loop formed a stable turn at neutral pH in wild-type PrP from both species. PrPSc-resistant mutations disrupted this turn even though only one of the mutants is in the X-loop. The X-loop is compact and buried in our previously described spiral models of PrPSc-like oligomers. On the basis of the findings presented here and in the context of the spiral oligomer model, we propose that expansion of the X-loop disrupts protofibril packing, providing a structural basis for resistance.
July | top
A Comparison of Multiscale Methods for the Analysis of Molecular Dynamics Simulations
Benson N.C. and Daggett V.
Journal of Physical Chemistry B 116:8722-8731, 2012
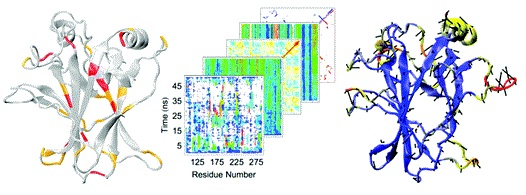
Molecular dynamics (MD) is the only technique available for obtaining dynamic protein data at atomic spatial resolution and picosecond or finer temporal resolution. In recent years, the cost of computational resources has decreased exponentially while the number of known protein structures, many of which are not characterized biochemically, has increased rapidly. These events have led to an increase in the use of MD in biological research, both to examine phenomena that cannot be resolved experimentally and to generate hypotheses that direct further experimental research. In fact, several databases of MD simulations have arisen in recent years. MD simulations, and especially MD simulation databases, contain massive amounts of data, yet interesting phenomena often occur over very short time periods and on the scale of only a few atoms. Analysis of such data must balance these fine-detail events with the global picture they create. Here, we address the multiscale nature of the problem by comparing several MD analysis methods to show their strengths and weaknesses at various scales using the wild-type and R282W mutant forms of the DNA-binding domain of protein p53. By leveraging these techniques together, we are able to pinpoint fine-detail and big picture differences between the protein.s variants. Our analyses indicate that the R282W mutation of p53 destabilizes the L1 loop and loosens the H2 helix conformation, but the loosened L1 loop can be rescued by residue H115, preventing the R282W mutation from completely destabilizing the protein or abolishing activity.
August | top
Structural and Functional Consequences of the Cardiac Troponin C L48Q Ca2+-Sensitizing Mutation
Wang D., Robertson I.M., Li M.X., McCully M.E., Crane M.L., Luo Z., Tu A.Y., Daggett V., Sykes B.D., and Regnier M.
Biochemistry51:4473-4487, 2012
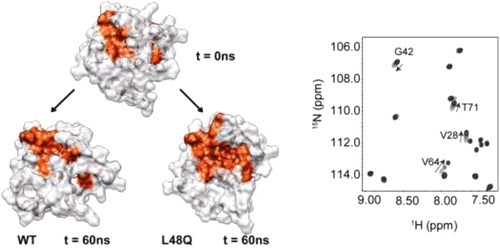
Calcium binding to the regulatory domain of cardiac troponin C (cNTnC) causes a conformational change that exposes a hydrophobic surface to which troponin I (cTnI) binds, prompting a series of protein.protein interactions that culminate in muscle contraction. A number of cTnC variants that alter the Ca2+ sensitivity of the thin filament have been linked to disease. Tikunova and Davis engineered a series of cNTnC mutations that altered Ca2+ binding properties and studied the effects on the Ca2+ sensitivity of the thin filament and contraction [Tikunova, S. B., and Davis, J. P. (2004) J. Biol. Chem. 279, 35341.35352]. One of the mutations they engineered, the L48Q variant, resulted in a pronounced increase in the cNTnC Ca2+ binding affinity and Ca2+ sensitivity of cardiac muscle force development. In this work, we sought structural and mechanistic explanations for the increased Ca2+ sensitivity of contraction for the L48Q cNTnC variant, using an array of biophysical techniques. We found that the L48Q mutation enhanced binding of both Ca2+ and cTnI to cTnC. Nuclear magnetic resonance chemical shift and relaxation data provided evidence that the cNTnC hydrophobic core is more exposed with the L48Q variant. Molecular dynamics simulations suggest that the mutation disrupts a network of crucial hydrophobic interactions so that the closed form of cNTnC is destabilized. The findings emphasize the importance of cNTnC.s conformation in the regulation of contraction and suggest that mutations in cNTnC that alter myofilament Ca 2+ sensitivity can do so by modulating Ca2+ and cTnI binding.
September | top
Promiscuous contacts and heightened dynamics increase thermostability in an engineered variant of the engrailed homeodomain.
McCully M.E., Beck D.A.C., Daggett V.
Protein Engineering & SelectionOnline:2012
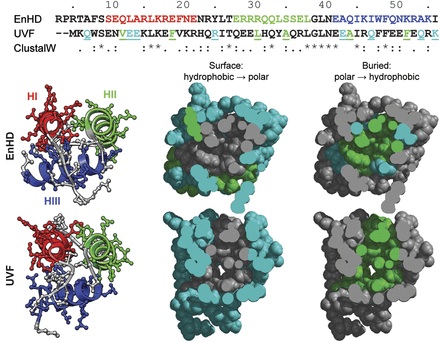
A thermostabilized variant (UVF) of the engrailed homeodomain (EnHD) was previously engineered by Mayo and co-workers. The melting temperature of the non-natural, designed protein is 50°C higher than the natural wild-type protein (>99 vs. 52°C), and the two proteins share 22% sequence identity. We have performed extensive (1 µs) all-atom, explicit solvent molecular dynamics simulations of the wild-type and engineered proteins to investigate their structural and dynamic properties at room temperature and at 100°C. Our simulations are in good agreement with nuclear magnetic resonance data available for the two proteins [nuclear Overhauser effect crosspeaks (NOEs), J-coupling constants and order parameters for EnHD; and NOEs for UVF], showing that we reproduce the backbone dynamics and side chain packing in the native state of both proteins. UVF was more dynamic at room temperature than EnHD, with respect to both its backbone and side chain motion. When the temperature was raised, the thermostable protein maintained this mobility while retaining its native conformation. EnHD, on the other hand, was unable to maintain its more rigid native structure at higher temperature and began to unfold. Heightened protein dynamics leading to promiscuous and dynamically interchangeable amino acid contacts makes UVF more tolerant to increasing temperature, providing a molecular explanation for heightened thermostability of this protein.