Paper of the Month | 2018
July | top
De Novo Designed Alpha-Sheet Peptides Inhibit Functional Amyloid Formation of Streptococcus Mutans Biofilms
Natasha Paranjapye, Valerie Daggett
Journal of Molecular Biology, Article in Press
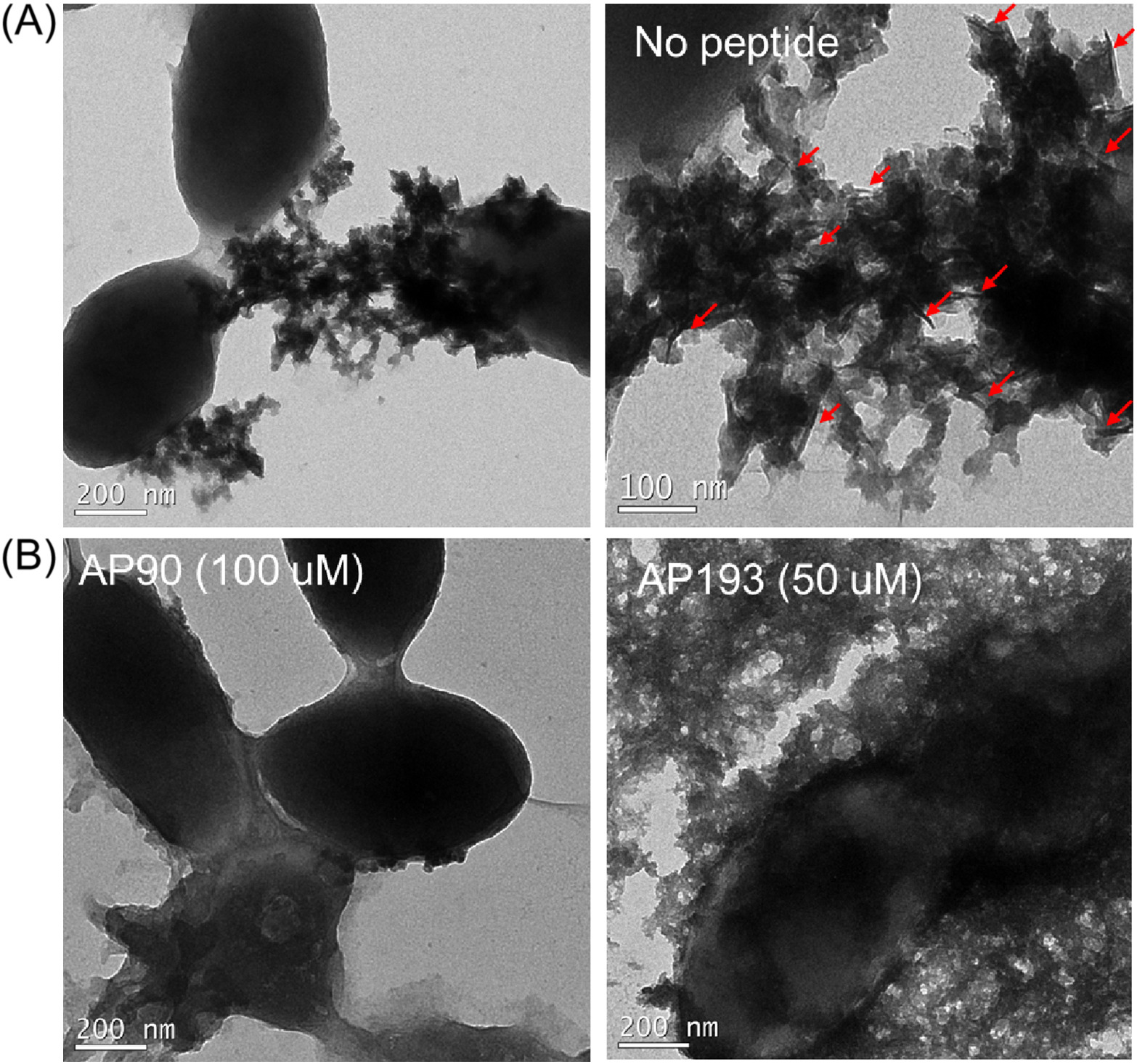
Streptococcus mutans is a bacterial species that predominates in the oral microbiome. S. mutans binds to the tooth surface, metabolizes sugars and produces acid, leading to cavity formation. S. mutans can also cause infectious endocarditis. Recent evidence suggests that S. mutans biofilms contain amyloid fibrils. Amyloids are insoluble fibrillar protein aggregates, and bacteria use functional amyloids to improve robustness of their biofilms. While the functional amyloids in bacteria such as E. coli and S. aureus have been heavily investigated, little is known about the mechanism of S. mutans amyloid formation. Previous results from our lab with the amyloidogenic proteins and peptides from the aforementioned bacteria and other mammalian amyloid systems suggest that amyloid formation progresses via an intermediate that adopts a unique secondary structure - α-sheet. De novo designed peptides with alternating L- and D- amino acid also adopt an α-sheet secondary structure and inhibit amyloid formation by binding to soluble oligomeric species during amyloidogenesis. Inhibition of fibrillization by α-sheet peptides suggests the presence of α-sheet during amyloid formation. To investigate the mechanism of functional amyloid formation in S. mutans, α-sheet peptides were compared to Epigallocatechin gallate (EGCG) for their ability to inhibit fibril formation in S. mutans. Inhibition was demonstrated in a biofilm plate assay and on hydroxyapatite surfaces both in S. mutans alone and in bacteria from human saliva. The observed inhibition suggests that an α-sheet mediated mechanism may be operative during functional amyloid formation.
September | top
Molecular dynamics-derived rotamer libraries for D-amino acids within homochiral and heterochiral polypeptides
Matthew Carter Childers, Clare-Louise Towse, Valerie Daggett
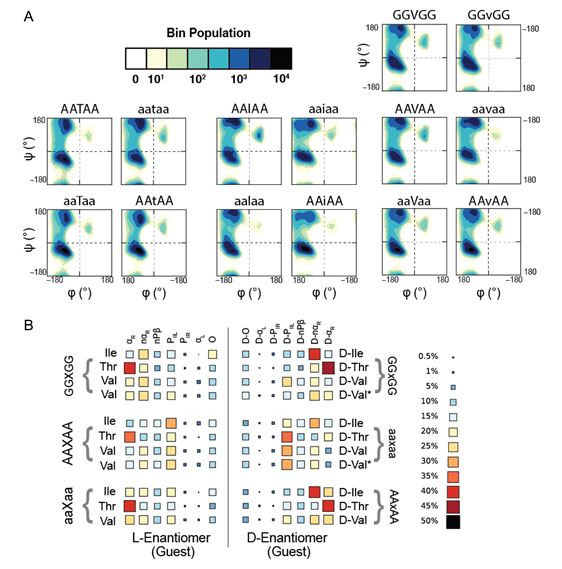
Computational resources have contributed to the design and engineering of novel proteins by integrating genomic, structural and dynamic aspects of proteins. Non-canonical amino acids, such as D-amino acids, expand the available sequence space for designing and engineering proteins; however, the rotamer libraries for D-amino acids are usually constructed as the mirror images of L-amino acid rotamer libraries, an assumption that has not been tested. To this end, we have performed molecular dynamics (MD) simulations of model host-guest peptide systems containing D-amino acids. Our simulations systematically address the applicability of the mirror image convention as well as the effects of neighboring residue chirality. Rotamer libraries derived from these systems provide realistic rotamer distributions suitable for use in both rational and computational design workflows. Our simulations also address the impact of chirality on the intrinsic conformational preferences of amino acids, providing fundamental insights into the relationship between chirality and biomolecular dynamics. While D-amino acids are rare in naturally occurring proteins, they are used in designed proteins to stabilize a desired conformation, increase bioavailability or confer favorable biochemical and physical attributes. Here, we present D-amino acid rotamer libraries derived from MD simulations of alanine-based host-guest pentapeptides and show how certain residues can deviate from mirror image symmetry. Our simulations directly model D-amino acids as guest residues within the chiral L-Ala and D-Ala pentapeptide series to explicitly incorporate any contributions resulting from the chiralities of neighboring residues.
October | top
Protein engineering reveals mechanisms of functional amyloid formation in Pseudomonas aeruginosa biofilms
Bleem, A., Christiansen, G., Madsen, D.J., Maric, H., Strømgaard K., Bryers, J.D., Daggett, V., Meyer, R.L., Otzen, D.E
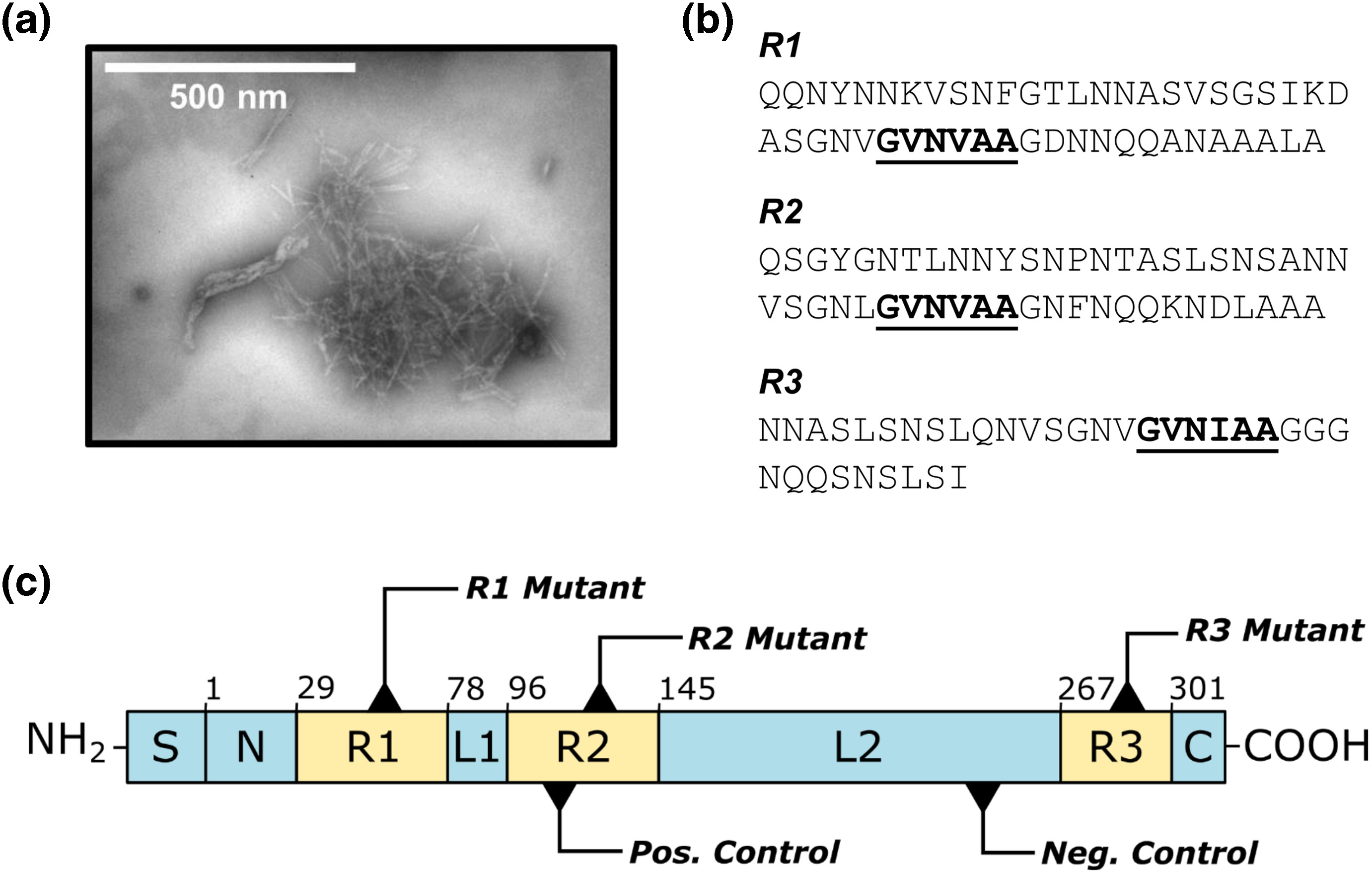
Amyloids are typically associated with neurodegenerative diseases, but recent research demonstrates that several bacteria utilize functional amyloid fibrils to fortify the biofilm extracellular matrix and thereby resist antibiotic treatments. In Pseudomonas aeruginosa, these fibrils are composed predominantly of FapC, a protein with high-sequence conservation among the genera. Previous studies established FapC as the major amyloid subunit, but its mechanism of fibril formation in P. aeruginosa remained largely unexplored. Here, we examine the FapC sequence in greater detail through a combination of bioinformatics and protein engineering, and we identify specific motifs that are implicated in amyloid formation. Sequence regions of high evolutionary conservation tend to coincide with regions of high amyloid propensity, and mutation of amyloidogenic motifs to a designed, non-amyloidogenic motif suppresses fibril formation in a pH-dependent manner. We establish the particular significance of the third repeat motif in promoting fibril formation and also demonstrate emergence of soluble oligomer species early in the aggregation pathway. The insights reported here expand our understanding of the mechanism of amyloid polymerization in P. aeruginosa, laying the foundation for development of new amyloid inhibitors to combat recalcitrant biofilm infections.