
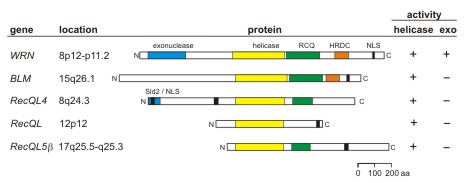
RecQ helicases
 Background:
The human RECQ helicases are 5-gene family that encodes proteins with a conserved
RECQ helicase consensus domain with 3'>5' DNA helicase activity. Mutations in the
WRN, BLM and RTS/RECQL4 genes lead to the genetic instability/ cancer
predisposition syndromes Werner syndrome, Bloom syndrome and Rothmund-Thomson
syndrome, respectively. Werner syndrome is the only RECQ helicase with an associated
exonuclease activity, and the only RECQ syndrome whose phenotype mimics premature
aging.
Background:
The human RECQ helicases are 5-gene family that encodes proteins with a conserved
RECQ helicase consensus domain with 3'>5' DNA helicase activity. Mutations in the
WRN, BLM and RTS/RECQL4 genes lead to the genetic instability/ cancer
predisposition syndromes Werner syndrome, Bloom syndrome and Rothmund-Thomson
syndrome, respectively. Werner syndrome is the only RECQ helicase with an associated
exonuclease activity, and the only RECQ syndrome whose phenotype mimics premature
aging.
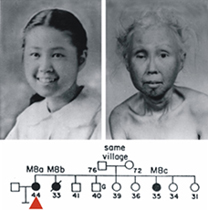
The two photos shown below are of a Japanese-American Werner syndrome patient as a teenager, and 30 years later as a woman in her mid-40's. This now-iconic photo pair shows the striking progeroid changes seen in Werner syndrome patients that begin in the 2nd decade of life. These changes are accompanied by an elevated risk of clinically important, age-associated diseases such as cancer, atherosclerotic cardiovascular disease, diabetes mellitus and osteoporosis. Patients may die prematurely in their 40's or 50's of either cancer or cardiovascular disease. A full description of the Werner syndrome phenotype can be found in recent publications from the lab, at the International Registry of Werner Syndrome website, or in the Online Mendelian Inheritance in Man (OMIM) listing for Werner syndrome at the National Library of Medicine.
WRN Mutation database: We also maintain the Werner Syndrome Locus-Specific Mutational Database for the Human Genome Organization and the Human Genome Variation Society. This database provides detailed information on WRN mutations identified in clinically ascertained Werner syndrome patients. The symbols in the figure below summarize the locations of mutations identified in Werner syndrome patients in the WRN protein and mRNA. The open and filled symbols in the figure below identify two large sets of mutations originally characterized by different investigators (see Mutation Database site for more detail).
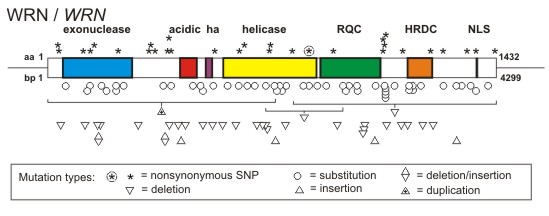
A key feature of all Werner syndrome-associated WRN mutations is that they truncate and promote the loss of WRN protein. This finding, together with functional phenotyping of WRN missense mutants that selectively lack exonuclease or helicase activity, indicate that the cellular phenotype of Werner syndrome develops only when both WRN catalytic activities are lost from cells. This loss also causes the clinical phenotype.
Current Research in the Monnat Lab: Our current research is focused on molecular functions of the RECQ helicases in genome stability assurance, DNA replication and the response to DNA damage.
DNA replication dynamics
Analysis of replicating molecules: We use cell-based, flow cytometric and single molecule methods to analyze DNA replication dynamics in human cells. These complementary methods provide mechanistic insight, and identify how genetic, environmental and pharmacologic perturbations that alter DNA replication dynamics.
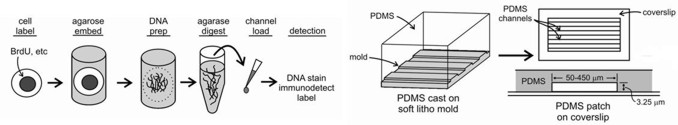
Microfluidic capillary channels and maRTA technology: Our core technology is maRTA, or microfluidic-assisted replication track analysis. Replicating DNA is labeled in vivo with halogenated pyrimidine nucleoside analogs such as bromodeoxyuridine (BrdU), and then isolated and stretched on glass for staining and fluorescent microscopy. Replicating DNA segments show up as linear immunostained tracks, and their lengths and distribution can be easily measured and quantified (see below). Sequential labeling with pairs of analogs (e.g. chloro- and iodo-deoxyuridine) provides a powerful way to reveal replication dynamics in vivo.
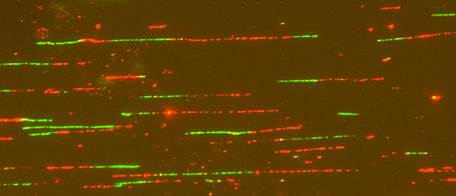
The use of microfluidic capillary channels in maRTA has helped solve the problematic step of reproducibly stretching replicating DNA molecules for immunostaining and quantification. Our microfluidic platform consists of parallel arrays of 3-sided, 3 or 4 µm high, 150-450 µm wide, and 1cm long capillary channels fabricated from polymethyl siloxane (PDMS) by conventional soft lithography, and silane-modified glass coverslips to reversibly seal the open side of the channels. Capillary tension in these microchannels facilitates DNA loading, stretching and deposition onto coverslip glass. A detailed discussion of the protocol and a step-by-step guide are available in our 2009 Nature Protocols Nature Protocols manuscript and related publications.
Genome Engineering
Genome Engineering:
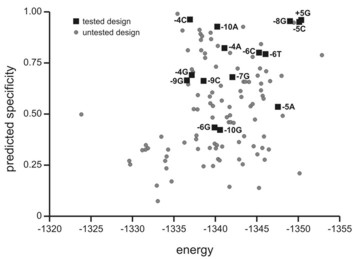
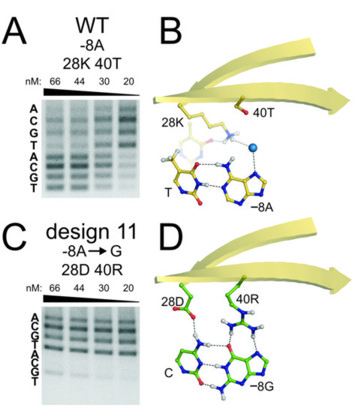
 We use three different nuclease platforms for genome engineering: homing endonucleases (so-called 'meganucleases'), TALENs and CRISPR/Cas9 nucleases. Our initial work in this area was based on the structural and biochemical characterization of several canonical homing endonuclease proteins, followed by their engineering to bind and cleave new target sites.
We use three different nuclease platforms for genome engineering: homing endonucleases (so-called 'meganucleases'), TALENs and CRISPR/Cas9 nucleases. Our initial work in this area was based on the structural and biochemical characterization of several canonical homing endonuclease proteins, followed by their engineering to bind and cleave new target sites.
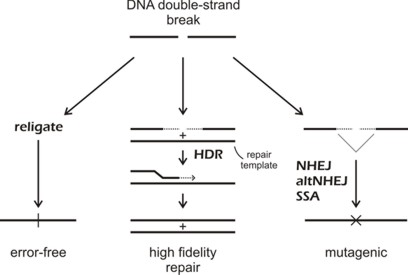
We have used these nucleases for a wide range of genome engineering projects: analyses of site-specific DNA double strand break dynamics [link to refs]; the targeted correction of heritable mutations giving rise to human bone marrow failure syndromes; and most recently engineering of the insect vector transmitting malaria, Anopheles gambiae, to interrupt malaria transmission.
These applications are part of highly collaborative research efforts developed as part of the Northwest Genome Engineering Consortium (NGEC), a U54 Interdisciplinary Research Consortium funded by the NIH Director's Fund; and as part of a Grand Challenges in Global Health award from the Gates Foundation that is administered by the Foundation for the NIH.
Human somatic mutation
We have used two different systems, the X-linked HPRT and ch.4 glycophorin-A (GPA) systems to quantify mutation frequencies, rates and molecular mutation spectrum in human cells and in vivo. We used both systems to characterize mutation frequency (and, in the case of HPRT, mutation rates and molecular mutation spectrum), in cells from patients with Werner syndrome. Although we no longer work actively on HPRT or GPA mutagenesis, we have included our most useful protocols and data to aid others working in this important area (see Links for protocols and data).
We also developed methods to isolate and analyze HPRT mutagenesis in primary tubular epithelial cells from human kidney. These analyses showed that spontaneous HPRT mutations were remarkably common in human kidney (plate photos below at left), and included a high proportion of new mutations and multiple mutations that had not been previously documented in human somatic cells. The frequency of HPRT mutants in kidney as a function of donor age is shown below right, together with comparison data from human peripheral blood T-lymphocytes.
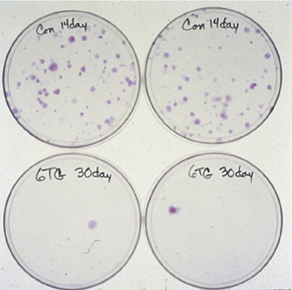
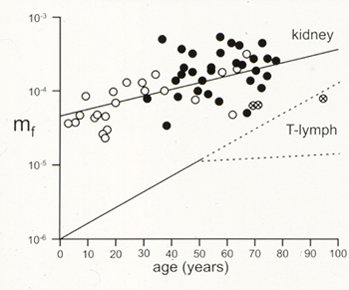
Our HPRT mutation analysis in human kidney was combined and analyzed with two additional large human HPRT mutation data sets: peripheral blood T-lymphocyte HPRT mutations, and germ line HPRT mutations from Lesch-Nyhan syndrome or gout patients. These mutation data sets are available as a spreadsheet for further analysis.