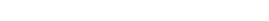
Higher-Order Protein Structure
The majority of proteins perform their function as part of a protein complex. The functional unit in these cases is not the individual protein, but the complete complex. Thus, to understand its mechanism of action requires the structure of each protein – not alone – but within its native macromolecular environment; the protein complex. Traditional methods of structural determination such as X-ray crystallography and NMR frequently encounter insurmountable obstacles when applied to large, multi-subunit protein complexes. Cryo-EM overcomes many of these obstacles; however, proteins must be relatively concentrated and cannot be present in crude cellular matrices. Furthermore, any protein-protein interactions to be observed must be stable enough to withstand purification and preparation.
The YRC is building on its previous achievements and developing technology described below to overcome these limitations and elucidate the structure of native protein complexes, their interaction interfaces, and their dynamics.
Cross-linking
 Model comparisons of the γTubulin small complex from Greenberg et al. (2016).
Model comparisons of the γTubulin small complex from Greenberg et al. (2016).Chemical cross-linking combined with mass spectrometry analysis (XL-MS) provides atomic level structural information on proteins and protein complexes. It is very rapid, requires little sample (a few micrograms of protein is sufficient), and purity is not of paramount importance. Furthermore, transient interactions are stabilized by the cross-linking process and are thus preserved. XL-MS does not provide a complete structure of the target protein. Instead, the method identifies pairs of residues within proteins and protein complexes that are within a specific distance from each other. Such information is highly complementary to structural information derived from other methods.
Molecular Painting
Cross-linking of protein complexes defines regions of proteins that interact within ~15 angstroms. As a complement to cross-linking, we are developing a “molecular painting” method to determine the regions of a protein complex that are solvent exposed and those which are not and thus potentially at interacting surfaces of a complex.
Co-evolution and Deep Mutational Scanning
We are developing technology to integrate the cross-linking and molecular painting data, co-evolution information, and deep-mutational scanning data to generate robust models of complexes. For more background on how co-evolution information relates to protein structure, see Ovchinnikov S et al. (2014).