Credit: Boston University CTE Center
By Genevieve Wanucha
As published in Dimensions Magazine - Fall 2019
For 2 days in October 2018, in Atlanta, Georgia, a group of researchers gathered in a large conference room. There were 30 people – more if you counted those on speaker phone. The working group included international experts in brain imaging, clinical diagnosis, genetics, neuropathology, and neuropsychology, as well as the Director and other leaders of the National Institute on Aging’s Alzheimer’s Disease Research Centers Program. They were there to discuss a longstanding conundrum in Alzheimer’s research. It was time to acknowledge that the field has crossed a tipping point, beyond which it is no longer possible to study brain health, Alzheimer’s disease, or memory loss without paying attention to a protein called TDP-43.
TDP-43 protein (TAR DNA binding protein-43) is critically important for normal nervous system function, but it can undergo chemical changes and go rogue in the context of brain disease. It’s bad news for brain cells. Abnormal clumps of TDP-43 are the hallmark pathology of amyotrophic lateral sclerosis (ALS) and half of frontotemporal lobar degeneration (FTLD) cases. It also can appear in other neurodegenerative diseases, including Lewy body dementia, Huntington’s disease, and chronic traumatic encephalopathy.
Then there’s the interesting and surprising occurrence of TDP-43 in the oldest old – the 80- and 90-year-olds who are diagnosed with Alzheimer’s disease or memory loss and those who are not. Consider the emerging consensus from a large community-based autopsy study that TDP-43 pathology is found in 1 in 5 individuals over 80 years. About a fifth of the oldest old have severe TDP-43 associated with episodic memory loss, which mimics the symptom profile of classic Alzheimer’s-type dementia.
Dr. C. Dirk Keene, MD, PhD leads the UW ADRC Neuropathology Core and studies TDP-43.
Researchers at many centers, notably Rush University, Mayo Clinic in Jacksonville, Florida, the University of Kentucky, and the UW ADRC, have looked harder and in more brain areas for TDP-43 in their aging cohorts. They have found that, more often than not, TDP-43 is an additional microscopic finding in cases of classic Alzheimer’s disease (defined as the presence of amyloid beta plaques and tau tangles) in older age groups. Most strikingly, TDP-43 pathology appears to hasten the cognitive decline in patients with co-existing Alzheimer’s pathology.
“So, we got together in Atlanta because we really felt strongly that researchers and clinicians should know that we are finding something else in the brain that seems to be very important,” said Dr. Julie Schneider, MD, MS, Professor of Pathology at Rush University and a leader of TDP-43 research, who spoke at the ADRC’s 2019 MODEL-AD Conference. “As our population ages, the landscape of what we are trying to treat is also going to change. The field needs to recognize that memory loss or dementia in older people may very well not be Alzheimer’s disease, as traditionally defined.”
This issue is significant news in medicine and public health, but it’s actually not a new discovery in the neuropathology field or a newly anointed human disease. “TDP-43 in older brains is something that neuropathologists have known about since 1993,” says Dr. C. Dirk Keene, MD, PhD, Associate Professor in UW Pathology and member of the consensus working group meeting. “But the condition hasn’t had a name and we’ve never had a way to talk about it. The idea of the meeting was that if we could come up with a common language around TDP-43, then at least we could communicate to each other and the public and have a common protocol for research around the country.”
Some Alzheimer’s researchers say it’s too early to issue guidelines and definitions around TDP-43 in older brains, before the field truly understands the details such as how TDP-43 in older brains is different than the TDP-43 in FTLD (typically a younger onset dementia). Yet, the working group in Atlanta decided that it’s past time to issue some definitive statement on the issue. At the meeting, they agreed that until specific biomarkers for TDP-43 exist, clinical trials will need to be designed to account for TDP-43 proteinopathy. For example, if drugs to treat Alzheimer’s pathology are tested in patients who also have TDP-43 in their brains, then the treatment effect might be diluted, so a larger number of participants should be enrolled.
A New Understanding of Brain Resilience
The UW ADRC has been studying TDP-43 at an accelerating pace in all autopsied brains over the past few years. Led by Dr. Caitlin Latimer, MD, PhD, Assistant Professor in UW Pathology, the ADRC has added nuance to the understanding of what TDP-43 actually means for the brain health of older adults. In a study published in Acta Neuropathologica Communications this year, Latimer found a strong correlation between the presence of TDP-43 pathology in the brain and those diagnosed with dementia in life.

Dr. Caitlin Latimer, MD/PhD, Assistant Professor in UW Pathology
Her study, which used autopsy brain tissue resources from the Adult Changes in Thought study, puts a special twist on the concept of resilience. ‘Resilience’ is the term for a person who had a high level of pathology in the brain, yet who stayed cognitively intact until death; while ‘resistance’ is a person who never develops brain pathology at all. In Latimer’s study, she showed that the brains of older people who had been resilient or resistant to Alzheimer’s disease pathology did not show TDP-43 pathology; yet almost all of the brains from age-matched people affected by symptoms of dementia during life did have TDP-43 pathology. Her findings give independent support to earlier work out of the Mayo Clinic showing that resistance to TDP-43 may be a big part of resilience to Alzheimer’s pathology.
Latimer goes as far as to suggest that retaining cognitive health late into life may very well depend on simply being resistant to developing TDP-43 pathology.
“When I finished this study, I had so many questions,” says Latimer. “Do TDP-43 and tau interact to cause disease? Is there a relationship in space and time between TDP-43 and Alzheimer’s disease pathology in the brain? What genetic factors confer resilience or resistance to these different pathologies?” These are questions that require supplementing the study of postmortem brain tissue with innovative techniques in molecular biology, such as modeling the toxic protein pathways in worms, mice, and human induced pluripotent stem cells models.
Insights from TDP-43 Experts
Dr. Nicole Liachko, PhD, Research Assistant Professor in UW Division of Gerontology & Geriatric Medicine, entered the field as a scientist studying neurodegeneration around the same time that TDP-43’s toxic effects were becoming known in ALS and FTLD in 2006. “I haven’t been surprised to see researchers finding TDP-43 in multiple neurodegenerative diseases including Alzheimer’s,” she says. “I’ve been following that aspect of the field since the beginning. I’ve been excited about it for 10 years, and finally everyone else is excited too.”
The Liachko Lab uses C.elegans worm models of human ALS, FTLD, and Alzheimer’s to study important aspects of the diseases. Worms are valuable tools in Alzheimer’s research because they have transparent bodies and simple nervous systems, which allow researchers to observe physiology in living detail. In recent worm model work, Drs. Liachko, Latimer, and Brian Kraemer, PhD, Research Associate Professor in UW Division of Gerontology & Geriatric Medicine, observed that tau and TDP-43 together make disease symptoms and progression much worse in the worms, even worse than would be expected if effects of tau and effects of TDP-43 were just added together. This finding may help explain why humans with both pathologies experience accelerated.
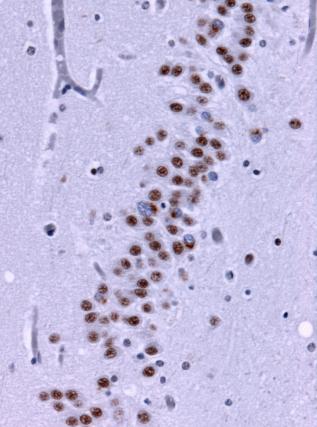
Under the microscope, brain tissue from a person with frontotemporal lobe degenerations shows deposits of TDP-43 (brown). Wikimedia Commons.
However, worm models are an artificial system; Liachko notes that the findings are only a jumping off point to study human biology. “In humans with Alzheimer’s disease, in neurons that show pathology, some will have tau aggregates, and others will have TDP-43. So, the question is, how are the neurons with TDP-43 making the tau pathology worse? Is it via synaptic connections, or through support cells in glia? It’s a fascinating question.”
Liachko’s team uses a technique that interferes with gene expression called RNAi. With this method, researchers can reduce the expression of individual genes in a worm’s genome and watch what happens as consequence. They search for ‘enhancers,’ or genes for which if you reduce their expression, it worsens symptoms, as well as ‘suppressors,’ or genes for which if you reduce expression, it improves things.
“What we have learned is that there are a number of cellular pathways that influence TDP-43 for better or worse, suggesting that there may be more than one way to intervene and improve TDP-43 toxicity,” says Liachko. “If we can figure out which is easiest to target and has the least side effects in cells, then we can hopefully pursue a treatment.”
But how does healthy TDP-43 become toxic in the first place?
Dr. Paul Valdmanis, PhD, Assistant Professor in UW Medical Genetics and ADRC collaborator, has an interest in TDP-43 from his background in the genetics of familial ALS and FTLD and helped to identify pathological variants in TDP-43 and TDP-43-related mechanisms as a major component of the diseases. He has insights into how healthy TDP-43 transforms into a problem for brain cells in ALS and FTLD, and perhaps different neurodegenerative conditions.
Healthy TDP-43 is what is called an ‘RNA-binding protein’, which means that it helps strands of RNA in the cell get translated into useful proteins. To fulfill this purpose, TDP-43 searches for particular sequences of RNA or other proteins floating around the brain cell, and it has affinity for certain ones.
“TDP-43 pathology appears across many diseases most likely because it is associating with other toxic proteins or substances aggregating at the same time,” he says. Valdmanis thinks that, in the brain cells of a person already developing toxic aggregations, TDP-43 still tries to do its normal job – but it runs into big trouble. “Maybe there is something within the pre-existing toxic inclusions that is sending out a signal for TDP-43 to associate,” he says. When TDP-43 reaches the destination, it finds a hornet’s nest and transforms into a problem itself.
The science of TDP-43 and neurodegenerative disease is still young and full of unanswered questions. Valdmanis wants to know the critical RNA or protein targets that TDP-43 naturally associates with, and how that interaction leads to aggregations in the setting of disease. “If we could figure out what that intermediary signal is, between TDP-43 and its biological targets, we could work to try to prevent that association and break down the propensity of some of these proteins to aggregate,” says Valdmanis. “We could precisely target the aberrant process central to a disease, without hindering all of the other targets that TDP-43 is supposed to interact with normally.”
In the field of medical genetics and behavioral neurology, TDP-43 has united ALS researchers and FTLD researchers, particularly since the 2011 discovery of a genetic expansion that causes both diseases. Now, new genetic treatments in development for ALS patients could also help prevent or delay forms of familial FTLD. “Perhaps advances in TDP-43 science in FTLD-ALS fields can benefit progress in Alzheimer’s disease, and vice versa,” says Valdmanis.
It’s never too LATE
The implications of the field’s evolving conversation are enormous and there are wide gaps in knowledge and diagnosis, but the tectonic plates of Alzheimer’s disease research have already shifted. At a public forum in January 2019 offered by the UW Institute for Stem Cell & Regenerative Medicine, a panel of ADRC researchers fielded questions from a curious audience after learning about multiple pathologies of dementia. “Is studying amyloid worth it anymore? Is the era of pursuing treatments for amyloid over?” they asked. Keene disagreed. “It’s not that we are abandoning amyloid, which is definitely a key player in Alzheimer’s disease; rather, TDP-43 gives us another target.” He pointed out the possibility that if a therapeutic could get rid of amyloid in the brain, then perhaps TDP-43 would never develop, so the field needs to understand the relationship between amyloid, tau, and TDP-43.

The panel for Repairing the Failing Brain (from left to right): Brad Rolf, M.S., CGC, Dr. Lorenz Studer, Dr. Jessica Young, Dr. C. Keene, and Dr. Eric Larson. WATCH the full video recording of the Public Forum
The results of the Atlanta working group were published in the journal Brain in 2019. Because TDP-43 pathology can occur on its own, independent of Alzheimer’s pathology, and because it looks to have a specific pattern of deposition and spread in the brain, researchers determined that TDP-43 in older brains is a distinct disease entity, named LATE (limbic-predominant age-related TDP-43 encephalopathy). The term LATE is primarily for use in the research world, to give guidance on how to characterize TDP-43 in research studies. Keene emphasizes that the new guidelines are only a basic minimum for ADRCs, and that individual centers can go further with innovative methods of TDP-43 research.
So what does this mean for patients? Currently, there is no way to detect TDP-43 in the brain during life, so no one can get a clinical diagnosis of LATE until biomarker tests are available. For now, neurologists are left with a process of elimination. “Doctors should suspect LATE if a patient screens negative on an amyloid or tau PET scan,” says Dr. Nina Silverberg, PhD, of the National Institute on Aging’s Alzheimer’s Disease Research Centers Program. Yet, few patients can obtain these specialty scans. Some worry that people diagnosed with Alzheimer’s will be confused as to whether they have the correct diagnosis, while doctors don’t have the tools to help clarify.
But Keene and researchers at the UW ADRC think that the effort to put a name and definition to LATE will benefit the public in the long run, especially when biomarker tests for amyloid, tau, and TDP-43 reach clinical usage. “The fact is that age-related cognitive decline can be caused by many things. At least we are forming a way to talk about this reality with our doctors and in the media,” he says. “I want a future where I can go to the doctor, and for them to say, ‘You have a build-up of amyloid and some tau, but no TDP-43 or vascular damage. And here’s a new treatment that may work for you.’ That’s the bottom line here.”
In light of this formal definition of LATE and the coming changes in the landscape of dementia prevention and treatment, the language we use to talk about dementia is requiring more and more nuance. “It’s important to distinguish between the pathology of Alzheimer’s disease – which is the presence of amyloid and tau in the brain – and the clinical syndrome,” says Dr. Thomas Grabowski, MD, Professor in UW Neurology and Radiology. “The term ‘Alzheimer’s-type dementia’ can serve as a placeholder term to describe a syndrome of memory loss that can have many underlying disease causes, including LATE.” In the robust conversation to continue, we should all remember that it’s never too late to ask questions and learn more about this rapidly changing field of science. •
__media-listing.png)


