
By Genevieve Wanucha and Franklin Faust
George Martin, MD, Emeritus Professor of UW Pathology, has led a long and productive career at the University of Washington, where he received his BS and MD degrees and has been a member of its faculty since 1957. Martin’s research has for many years been concerned with the development of genetic approaches to the study of aging and age-related diseases. At the UW, he is recognized for creating research opportunities in neuropathology and the biology of aging. By the time he founded the ADRC in 1985, he had already started the UW Medical Scientist Training Program in 1970 and the UW Genetic Approaches to Aging Research Institutional Training Grant, supported by the National Institute on Aging. At 93 years of age, George Martin is devoted to supporting research in aging and genetics and thinking critically about the most challenging issues in studying Alzheimer’s disease and related neurodegenerative conditions.
He shares memories, fascinating insights about evolutionary biological theories of aging and genetics, and his hopes for the field of Alzheimer’s disease research.
How did you first become involved with aging and genetics research?

Earl Benditt, MD in 1950. Courtesy, University of Chicago Library.
After I finished my medical residency at the University of Chicago, my late colleague and Associate Professor Earl Benditt, MD asked me to join him as a faculty member at the University of Washington in 1957. Benditt had been recruited to build an academic pathology department and jump-started research in the department on a modest National Institutes of Health grant.
I had developed an interest in the neurobiology of the genetic disorder called Wilson's disease in my medical residency, so I decided to connect with the chair of the Department of Medical Genetics and the late Professor Arno Motulsky, the founder of Medical Genetics here at the UW. I said: “I want to learn more about genetics!” They were very welcoming to me. I asked them to participate in our pathology course for the first time. So, I had to coordinate by bringing medical genetics experts in to give lectures to the new Department of Pathology.

At the time, I was working on an enzyme called caeruloplasmin, which is dysfunctional in Wilson's disease. Wilson’s disease prevents the body from getting rid of excess copper, and these patients develop copper deposits in their liver and around their eyes. That work led to the cloning of the copper-transporting gene, ATP7B. Defects in ATP7B remain the only known genetic cause for Wilson's disease. And I said, “Hey, that's interesting! What about the element vanadium? It's a close cousin of copper.” Our experiments indeed showed that vanadium made a tremendous difference in modulating organic compounds produced by the body, such as serotonin and adrenaline.
There was no predicting where that research would go. In a literature search, I came across the late Guido Pontecorvo, “Ponti.” He was the first person who suggested it was possible to map human genes based upon his research on mitotic recombination in a fungus called Aspergillus nidulans! So, I took my whole family to Scotland for the year of 1961, where my young daughter developed a brilliant Scottish accent! —I worked with Ponti at the University of Glasgow for the entire year. That same year, the now famous paper by Leonard Hayflick came out on the Hayflick limit, or the number of times a normal human cell population will divide before cell division stops. Hayflick showed that there's a limited lifespan of cells and there's some evidence that it's related to aging. That’s how I first got into aging research and genetics, and now I am trying to push research into how aging is the fundamental risk factor in all variants of Alzheimer's disease.

Arno Motulsky, MD in 1955
Why did you turn your attention to Alzheimer’s disease and decide to start the ADRC with a focus on genetics?
It was the late Carl Eisdorfer, MD who first interested me in Alzheimer’s disease at the UW. Eisdorfer started the Gerontological Research and Clinical Center at the VA Puget Sound Health Care System and served as chair of the UW Department of Psychiatry. He published the first UW paper on Alzheimer’s disease in 1980, reporting on the immune system in people with the disease.
Around that time, my friend and colleague Rudolph Tanzi, PhD [now the Director of the Genetics and Aging Research Unit at Massachusetts General Hospital] published the 1983 study titled, A polymorphic DNA marker genetically linked to Huntington’s disease. This linkage for Huntington's disease really turned me on to the power and possibility of linking a gene to a neurodegenerative disease. My colleague Thomas Bird, MD, who founded the UW Neurogenetics clinic in 1974, was seeing more and more families with a history of Alzheimer’s disease. So, we decided to apply to become an ADRC with the overarching theme of studying familial Alzheimer’s disease.

Guido Pontecorvo, FRS in 1975
I'm proud of what the original genetics team ultimately accomplished in the field of familial Alzheimer’s disease. Our group’s successful contributions came from individuals working collectively. Thomas Bird, Gerard Schellenberg, and Ellen Wijsman were key to our early work– as well as the post-doctoral fellows and many others. They made the contributions that we’re seeing emerging from our Center today possible.
What is a scientific mystery of Alzheimer’s disease genetics that still fascinates you?
The Apolipoprotein E gene (APOE) poses interesting questions, and it's very complex. We have known since 1993 that APOE ε4, one polymorphism of APOE, is a substantial risk factor for late-onset Alzheimer’s disease in the European population. Carrying an APOE ε4 allele doesn’t guarantee someone will develop Alzheimer’s, but the risk goes up circa three-fold with one allele and circa twelve-fold with two; the risky allele is present in about 25% of the human population.
I was impressed with the fact that APOE ε4 is actually the dominant and most common allele in non-human primates, who do not develop Alzheimer’s disease! I became interested in how the risky allele variant is harmful in some environments, but it may actually be beneficial in others for evolutionary reasons.

Joshua Lederberg, PhD in 1962
When I was on a sabbatical with the late Joshua Lederberg, PhD [at Rockefeller University], I pursued an idea I had. Lederberg was interested in infectious disease and molecular biology. And I said, “Josh, I think that maybe that the reason non-human primates have APOE ε4 is that it protects them from some infectious disease.” I said that because I knew that APOE codes for protein that is involved in transport of lipids in the blood and the central nervous system. I also knew of evidence that the APOE ε4 allele is not as effective at putting lipids into cell membranes, as are the APOE ε2 or ε3 alleles. Lederberg assured me that there were some fungal agents, such as T. brucei, that need the host lipids for them to survive because they can't synthesize the complex lipids on their own. So, if you—the infected host—have an APOE ε4 allele, then you wouldn’t make the amount of lipids needed by the pathogen, moderating or preventing the disease and leading to a longer life and increased reproductive potential for you.
Now, there is more evidence for this hypothesis. My friend Caleb Finch, PhD at the University of Southern California argues that there may sometimes be positive reasons for carrying APOE ε4 in terms of cognition. While being an APOE ε4 carrier is the strongest risk factor to date of late-onset Alzheimer’s and cognitive decline in industrial populations; it is associated with greater cognitive performance in individuals facing a high parasite and pathogen load, suggesting advantages to the APOE ε4 allele under certain environmental conditions.
In fact, there is evidence that children living in poor hygiene environments who carry the APOE ε4 variant are more resistant to diarrheal disease, including malaria, providing a survival advantage. This phenomenon may explain why some equatorial populations have a higher prevalence of this particular gene variant.
Remember, your risk for Alzheimer’s disease is not only about genes, and it's not only about environments. It's gene-environment interactions, and it's gene-gene interactions. Genes elsewhere in the genome can make a big difference to whether that mutation is going to get you in trouble or not. And we don't know enough about all those background genomes. That's going to be a tremendous task in the future.
What is your hope for the direction of the ADRC? If you could decide to put funding towards any area, what would it be?
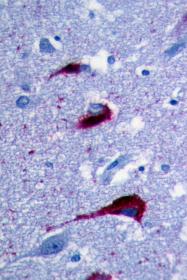
Neurofibullary tangles of tau protein in post-mortem brain tissue, as viewed under a microscope
I've been telling people my perspective for years, and even today not everybody agrees with me! I remember a moment at an international conference in 2009. In my talk, I said: “There are many different types of Alzheimer's, and this is a good heuristic notion.” A famous researcher responded: "Hey, George! There's only one type of Alzheimer's disease!” But I keep saying: Amyloid beta is important in Alzheimer’s disease. Tau protein is important. Chronic inflammation is important. But what happens before that? There may be different biological pathways in different people. Even infectious disease might relate to changes in the brain’s immune system, such as the herpes simplex virus, but it could be something else.
So, I would push for more work on the fundamental mechanisms of aging. Aging mechanisms comprise a big pathway, though not the only pathway. My dream would be that one day we'll be able to address a person’s relative susceptibility to one of the numerous hallmarks of aging. For example, it would be useful to know if the person has robust retention of mitochondrial function, or if they have comparatively low rates of somatic mutation hallmarks of aging. After death, that estimate would give more meaningful context to the neuropathological findings from brain autopsies, brain imaging data, and genomics. You could say: “Hey, this guy got in trouble with Alzheimer’s because of protein trafficking function.” You'd have that full picture of an individual’s risk and disease trajectory, but we don't have the financial resources for this approach currently.
Coupled with that biological information, we should capture data about the individual’s background and environmental exposures in greater depth—for example, whether the person was exposed to a lot of toxins. We don't generally have that type of health information. We would need a universal health care electronic records system, so you could look up all those parameters for the question you’d like to answer. Let's hope we get there. I would love to see a research grant by the NIH supporting that kind of comprehensive dataset for research participants.
What is your own favorite hypothesis?
Epigenetic gambling. The best example comes from studies of wild-type Caenorhabditis elegans worms when grown in sterile suspension cultures. Their genes are identical, like identical twins. The environment is identical. In those experiments, biogerontologists have observed for many decades that, despite all efforts to control genotype and environment, there are substantial variations in the lifespan of individual worms. In fact, the expression of genes in a family of identical cells gets more and more different as an organism gets older. I raised a hypothetical mechanism for this observable variation: Nature wants to gamble with gene expression. I call that ‘epigenetic gambling’.
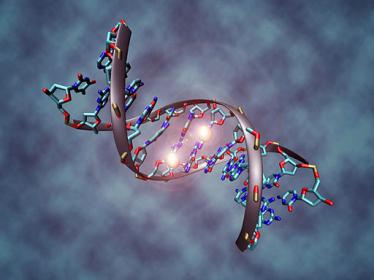
This image shows a DNA molecule that is methylated on both strands on the center cytosine. DNA methylation plays an important role for epigenetic gene regulation in development and cancer. Christoph Bock, Max Planck Institute for Informatics
In epigenetic gambling, random changes in cellular gene expression evolved as an adaptive mechanism to ensure survival of members of a group in the face of unpredictable environmental challenges. Once activated, it could lead to ‘epigenetic drift’, or changes in gene activity with aging, amongst all members of the group. I predict that while particular patterns of gene expression will be adaptive for a subset of individuals within a population early in life, continued epigenetic drift will result in variable patterns of disease over time. The weakness of this hypothesis is that we do not currently have a plausible molecular mechanism for the genetic ‘randomizer’ of epigenetic gene expression.
My friend Dan Promislow, PhD [UW Department of Laboratory Medicine and Pathology] has informed me there's a name for epigenetic gambling in evolutionary biology. It's called ‘bet-hedging,’ a concept which is very much in concert with my notion of epigenetic gambling.
And that's what I would like to test in Alzheimer's disease and other disorders in model systems. New technology to study gene expression in the brain is just exciting as hell. Jay Shendure, MD, PhD [UW Genome Sciences] can do what we call single-cell transcriptomes. You can see what's happening in the cells in the early stages of Alzheimer’s disease, during the screw-up in the proteins that are transcribed. I wish I were a young man again with this technology. It's just unbelievable what you can do.
What is your hope for the future of brain health and disease prevention?
If I had a lot of money and power, I would put a tremendous amount of money into early education. And it's the single most desperate thing that we need to do now: address inequality in education and the rampant misunderstanding of scientific evidence. Correlation is not causation. That's where we should put our money. I love what the mayor of New York City is doing. Mandatory education, preschool, beginning at age 3. The key is, you've got to give these teachers a lot more money, and you've got to train them in teaching critical thinking. Kids have curious little minds. And when they see something new or hear about something new, they’ll say, “Gee, teacher, that's interesting.” The teacher should then instruct them to automatically ask: How do we know that's true? Can you imagine if every kid grew up in our democracy asking the question: “How do we know that's true? What is the evidence?” It would be a transformation.
You are 93 years old, regularly attend seminars, co-author journal articles and keep up with the latest advances in research. What do you attribute to this incredible super-aging?

Your question is one that deserves a great deal of research. While we have gotten a good start on learning a bit about the phenomena of resistance and resilience to basic mechanism of aging and diseases of aging via the study of unusually healthy centenarians, we need more research on what I have named Unimodal Antigeroid Syndromes—the genetic and environmental basis for resistance and resilience to each of the numerous specific age-related disorders.
In my case, my number one hypothesis is good luck. I probably inherited a few good genetic variants, which I may share with a beloved Uncle who remains cognitively sharp and who will shortly become a centenarian. I also was lucky enough to have gotten outstanding teachers and fellow students from early primary school to postdoctoral training. On the other hand, when my Mom asked me if I wanted to pursue the option of attending kindergarten, my answer was “No, Mom, I want to have one more year to sleep late!” •


_(cropped)__full__media-listing.jpg)

__media-listing.png)